Disorders of Hemostasis I: Coagulation
![]()
APPROACH TO THE BLEEDING PATIENT
Abnormalities of the activity of coagulation proteins and related molecules, decreased platelet function, or disruption of the vasculature (as by surgery or trauma) can lead to bleeding. Careful assessment of both the clinical history and laboratory testing are necessary to establish the reason for bleeding.
![]() Initial laboratory studies in a patient with new-onset or recent bleeding include a platelet count, activated partial thromboplastin time (aPTT), prothrombin time (PT), and fibrinogen. If the bleeding is moderate to severe, a hemoglobin level and specimen for red cell cross-matching should also be sent.
Initial laboratory studies in a patient with new-onset or recent bleeding include a platelet count, activated partial thromboplastin time (aPTT), prothrombin time (PT), and fibrinogen. If the bleeding is moderate to severe, a hemoglobin level and specimen for red cell cross-matching should also be sent.
![]() The character, timing, and location of the bleeding should be considered. Is the bleeding spontaneous, or associated only with invasive procedures or trauma? If peri-procedural, is the bleeding immediate or delayed? Mucocutaneous bleeding (epistaxis, gingival hemorrhage, petechiae/ecchymoses, gastrointestinal, and urinary tract bleeding) are more characteristic of a defect in the activity of platelets, whereas soft-tissue bleeding or hemarthrosis suggests a deficiency in the activity of coagulation factors.
The character, timing, and location of the bleeding should be considered. Is the bleeding spontaneous, or associated only with invasive procedures or trauma? If peri-procedural, is the bleeding immediate or delayed? Mucocutaneous bleeding (epistaxis, gingival hemorrhage, petechiae/ecchymoses, gastrointestinal, and urinary tract bleeding) are more characteristic of a defect in the activity of platelets, whereas soft-tissue bleeding or hemarthrosis suggests a deficiency in the activity of coagulation factors.
![]() The clinical context is very important in determining the reason for bleeding. Hemorrhage in a patient who has been receiving heparin or warfarin may indicate excess anticoagulation or presence of a previously undetected lesion. A lifelong history of excessive bleeding may occur due to an inherited disorder of hemostasis. Bleeding in an individual who is in septic shock may point to disseminated intravascular coagulation (DIC). New-onset, diffuse bleeding in a patient who is pregnant or post-partum may signify the HELLP syndrome or other entities. Postsurgical bleeding may stem from a number of causes, but an initial consideration should be deficient hemostasis due to a traumatized, bleeding vessel, as well as a coagulation factor defect or deficits.
The clinical context is very important in determining the reason for bleeding. Hemorrhage in a patient who has been receiving heparin or warfarin may indicate excess anticoagulation or presence of a previously undetected lesion. A lifelong history of excessive bleeding may occur due to an inherited disorder of hemostasis. Bleeding in an individual who is in septic shock may point to disseminated intravascular coagulation (DIC). New-onset, diffuse bleeding in a patient who is pregnant or post-partum may signify the HELLP syndrome or other entities. Postsurgical bleeding may stem from a number of causes, but an initial consideration should be deficient hemostasis due to a traumatized, bleeding vessel, as well as a coagulation factor defect or deficits.
![]() A family history of bleeding raises the clinical suspicion for heritable disorders such as hemophilia A or B (X-linked recessive inheritance) or von Willebrand disease (VWD) (autosomal dominant inheritance in most cases).
A family history of bleeding raises the clinical suspicion for heritable disorders such as hemophilia A or B (X-linked recessive inheritance) or von Willebrand disease (VWD) (autosomal dominant inheritance in most cases).
![]() Importantly, bleeding does not necessarily indicate an intrinsic abnormality of hemostasis. Individuals with normal coagulation and platelet function will bleed given a sufficient hemostatic challenge (trauma, surgery, invasive malignancy).
Importantly, bleeding does not necessarily indicate an intrinsic abnormality of hemostasis. Individuals with normal coagulation and platelet function will bleed given a sufficient hemostatic challenge (trauma, surgery, invasive malignancy).
![]()
Coagulation Factors: Background
![]() Coagulation factors (clotting factors) are synthesized in the liver.
Coagulation factors (clotting factors) are synthesized in the liver.
![]() Factors II, VII, IX, X, XI, and XII are serine proteases that are inactive as synthesized and acquire enzymatic capability when cleaved (activated) by other proteins. A post-synthetic step in the production of factors II, VII, IX, and X and the natural anticoagulant proteins C and S requires the activity of a vitamin K–dependent carboxylase that modifies the amino-terminus of each factor, enabling it to function.
Factors II, VII, IX, X, XI, and XII are serine proteases that are inactive as synthesized and acquire enzymatic capability when cleaved (activated) by other proteins. A post-synthetic step in the production of factors II, VII, IX, and X and the natural anticoagulant proteins C and S requires the activity of a vitamin K–dependent carboxylase that modifies the amino-terminus of each factor, enabling it to function.
![]() Tissue factor (TF) and factors V and VIII serve as cofactors for coagulation reactions.
Tissue factor (TF) and factors V and VIII serve as cofactors for coagulation reactions.
![]() The activity of all clotting factors culminates in a principal event: the generation of thrombin at sites of vascular injury. Thrombin activates platelets (primary hemostasis) and cleaves fibrinogen to form fibrin (secondary hemostasis) at sites of blood vessel compromise.
The activity of all clotting factors culminates in a principal event: the generation of thrombin at sites of vascular injury. Thrombin activates platelets (primary hemostasis) and cleaves fibrinogen to form fibrin (secondary hemostasis) at sites of blood vessel compromise.
![]() The normal laboratory range of factor activity levels is ˜50% to 150% and is derived from plasma activity as observed in a reference pool of normal donors. The hemostatic level of a given clotting factor (the level of factor necessary to maintain normal hemostasis) typically is much lower. For instance, 5% activity of factor VIII is well below the laboratory reference range but usually is sufficient to prevent spontaneous bleeding.
The normal laboratory range of factor activity levels is ˜50% to 150% and is derived from plasma activity as observed in a reference pool of normal donors. The hemostatic level of a given clotting factor (the level of factor necessary to maintain normal hemostasis) typically is much lower. For instance, 5% activity of factor VIII is well below the laboratory reference range but usually is sufficient to prevent spontaneous bleeding.
The Coagulation Cascade
The coagulation cascade illustrates the activation of coagulation factors in the formation of a fibrin clot. It comprises the tissue injury (also known as extrinsic), contact (also known as intrinsic), and common pathways of coagulation (Fig. 20.1). The pathways within the coagulation cascade probably best reflect the activity of clotting factors in vitro, whereas in vivo the pathways not only interact at multiple points but also function in concert with the activation and aggregation of platelets to achieve hemostasis.
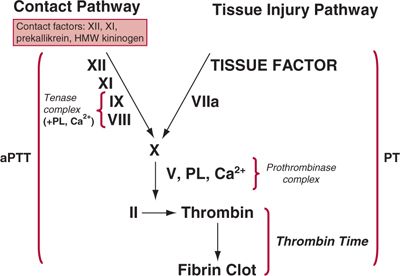
FIGURE 20.1. The coagulation cascade.The tissue injury pathway of coagulation begins with the binding of activated factor VII (VIIa) to tissue factor (TF), which is provided by cell membranes.VIIa converts X to Xa.The prothrombinase complex, formed by the binding of Xa to Va in the presence of PL and Ca2+, converts II (prothrombin) to IIa (thrombin).The contact pathway of coagulation begins with the activation of factor XII to XIIa by kallikrein. XIIa cleaves XI to XIa; XIa cleaves IX to IXa. IXa forms a complex with VIIIa in the presence of PL and Ca2+ (tenase complex), and converts X to Xa. Xa, in the presence of Va, PL, and Ca2+, then cleaves II (prothrombin) to IIa (thrombin).The common pathway involves the cleavage of II by prothrombinase to yield thrombin, and thrombin cleavage of fibrinogen to form fibrin, which is then cross-linked via the action of XIIIa. Activation of coagulation usually begins with the tissue injury system, which provides feedback to the contact system by IIa-mediated activation of factor XI.There are additional points of interaction between the pathways (not indicated).
![]() Tissue injury pathway. The tissue injury pathway of coagulation begins with the binding of activated factor VII (VIIa) to TF. The TF-VIIa complex mediates the conversion of X to Xa. The prothrombinase complex, formed by the binding of Xa to Va on a phospholipid (PL) surface (usually platelet membranes) in the presence of Ca2+, converts II (prothrombin) to IIa (thrombin).
Tissue injury pathway. The tissue injury pathway of coagulation begins with the binding of activated factor VII (VIIa) to TF. The TF-VIIa complex mediates the conversion of X to Xa. The prothrombinase complex, formed by the binding of Xa to Va on a phospholipid (PL) surface (usually platelet membranes) in the presence of Ca2+, converts II (prothrombin) to IIa (thrombin).
![]() Contact pathway. Activation of contact factors at the site of vascular injury leads to the conversion of Factor XII to XIIa, and the sequential conversion of XI to XIa and IX to IXa. IXa complexes with VIIIa, PL, and Ca2+, forming the tenase complex, which converts X to Xa. Xa, in a complex with Va, PL, and Ca2+ then cleaves II (prothrombin) to IIa (thrombin). The TF-VIIa complex can also activate IX leading to the subsequent formation of the tenase complex.
Contact pathway. Activation of contact factors at the site of vascular injury leads to the conversion of Factor XII to XIIa, and the sequential conversion of XI to XIa and IX to IXa. IXa complexes with VIIIa, PL, and Ca2+, forming the tenase complex, which converts X to Xa. Xa, in a complex with Va, PL, and Ca2+ then cleaves II (prothrombin) to IIa (thrombin). The TF-VIIa complex can also activate IX leading to the subsequent formation of the tenase complex.
![]() Common pathway. The tissue injury and contact pathways converge in the common pathway, where X is converted to Xa, and prothrombin (II) is cleaved to form thrombin. Thrombin then cleaves fibrinogen to form fibrin, which is then cross-linked via the action of XIII.
Common pathway. The tissue injury and contact pathways converge in the common pathway, where X is converted to Xa, and prothrombin (II) is cleaved to form thrombin. Thrombin then cleaves fibrinogen to form fibrin, which is then cross-linked via the action of XIII.
Common Coagulation Tests
An understanding of the basic laboratory tests for coagulation assists in the evaluation of bleeding disorders.
![]() The PT is performed by adding thromboplastin (TP), composed of crude or recombinant TF plus Ca2+, to plasma that has been anti-coagulated with citrate, and the time to formation of a fibrin clot is measured. Because the PT comprises reactions of coagulation that occur in the tissue injury and common pathways of coagulation, deficiencies in the activity of II, V, VII, X, or fibrinogen may prolong the PT.
The PT is performed by adding thromboplastin (TP), composed of crude or recombinant TF plus Ca2+, to plasma that has been anti-coagulated with citrate, and the time to formation of a fibrin clot is measured. Because the PT comprises reactions of coagulation that occur in the tissue injury and common pathways of coagulation, deficiencies in the activity of II, V, VII, X, or fibrinogen may prolong the PT.
![]() The International Normalized Ratio (INR) was developed to standardize the reporting of PT values in warfarin-anticoagulated patients. Standardization is necessary because commercially available TP reagents have varying potencies that directly impact the PT; one TP may yield a different PT result than another when the same sample is tested. The potency of a given TP is expressed in terms of the International Sensitivity Index (ISI).
The International Normalized Ratio (INR) was developed to standardize the reporting of PT values in warfarin-anticoagulated patients. Standardization is necessary because commercially available TP reagents have varying potencies that directly impact the PT; one TP may yield a different PT result than another when the same sample is tested. The potency of a given TP is expressed in terms of the International Sensitivity Index (ISI).
![]() Because the INR was developed to report factors measured by the PT that are decreased by warfarin impairment of vitamin K–mediated synthesis (the INR is not standardized for abnormalities of factor V and fibrinogen), the INR should be used only to describe anticoagulation in patients who are receiving warfarin. In all other patients (such as patients with liver disease), the PT should be referenced.
Because the INR was developed to report factors measured by the PT that are decreased by warfarin impairment of vitamin K–mediated synthesis (the INR is not standardized for abnormalities of factor V and fibrinogen), the INR should be used only to describe anticoagulation in patients who are receiving warfarin. In all other patients (such as patients with liver disease), the PT should be referenced.
![]() The formula for the INR is (PTpatient/PTmean normal)ISI.
The formula for the INR is (PTpatient/PTmean normal)ISI.
![]() The aPTT begins with the addition of a contact activating agent to citrate-anticoagulated plasma. PL and Ca2+ are added, and the time for formation of a fibrin clot is measured. Because the aPTT reflects reactions of coagulation that occur in the contact and common pathways of coagulation, deficiencies in the activity of factors II, V, VIII, IX, X, XI, or XII may prolong the aPTT. A deficiency of other contact factors, such as prekallikrein or high molecular weight kininogen (HMWK), may also prolong the aPTT. Isolated abnormalities of fibrinogen rarely impact the aPTT.
The aPTT begins with the addition of a contact activating agent to citrate-anticoagulated plasma. PL and Ca2+ are added, and the time for formation of a fibrin clot is measured. Because the aPTT reflects reactions of coagulation that occur in the contact and common pathways of coagulation, deficiencies in the activity of factors II, V, VIII, IX, X, XI, or XII may prolong the aPTT. A deficiency of other contact factors, such as prekallikrein or high molecular weight kininogen (HMWK), may also prolong the aPTT. Isolated abnormalities of fibrinogen rarely impact the aPTT.
![]() The long-incubation aPTT is performed by incubating the sample with activating agents for 10 minutes prior to the addition of PL and Ca2+. If the contact factor prekallikrein is deficient, this extra incubation time allows activation of factor XII and correction of the aPTT.
The long-incubation aPTT is performed by incubating the sample with activating agents for 10 minutes prior to the addition of PL and Ca2+. If the contact factor prekallikrein is deficient, this extra incubation time allows activation of factor XII and correction of the aPTT.
![]() Mixing studies are performed using a mixture of 50% patient plasma and 50% normal control plasma; the PT or aPTT is then performed as usual. Correction of a prolonged PT or aPTT with mixing generally implies a qualitative or quantitative abnormality of one or more clotting factors in the patient plasma. In contrast, failure of the PT or aPTT to correct completely upon mixing suggests the presence of an inhibitor in the patient plasma that neutralizes a component of the patient and normal plasma. Both lupus anticoagulants (LAs; see below) and inhibitors to specific clotting factors can result in a prolonged aPTT or PTT that does not correct upon mixing.
Mixing studies are performed using a mixture of 50% patient plasma and 50% normal control plasma; the PT or aPTT is then performed as usual. Correction of a prolonged PT or aPTT with mixing generally implies a qualitative or quantitative abnormality of one or more clotting factors in the patient plasma. In contrast, failure of the PT or aPTT to correct completely upon mixing suggests the presence of an inhibitor in the patient plasma that neutralizes a component of the patient and normal plasma. Both lupus anticoagulants (LAs; see below) and inhibitors to specific clotting factors can result in a prolonged aPTT or PTT that does not correct upon mixing.
![]() When evaluating a prolonged aPTT, an aPTT is performed on the mixture, then the mixture is allowed to incubate for an hour and the aPTT is repeated; some inhibitors of factor VIII are maximally inhibitory at an hour or more post-mix. For instance, the aPTT on a 1:1 mixture of normal and patient plasma containing a factor VIII inhibitor may show correction initially but demonstrate a prolongation at 1 hour.
When evaluating a prolonged aPTT, an aPTT is performed on the mixture, then the mixture is allowed to incubate for an hour and the aPTT is repeated; some inhibitors of factor VIII are maximally inhibitory at an hour or more post-mix. For instance, the aPTT on a 1:1 mixture of normal and patient plasma containing a factor VIII inhibitor may show correction initially but demonstrate a prolongation at 1 hour.
![]() Occasionally, a weak LA may produce a prolonged aPTT or PT that corrects on mixing.
Occasionally, a weak LA may produce a prolonged aPTT or PT that corrects on mixing.
![]() The bleeding time (BT) involves making a controlled incision in soft tissue (usually at a site on the forearm) and measuring the time to cessation of bleeding. Anemia and abnormalities of coagulation factors, platelets, or the vasculature may prolong the BT. The BT does not correlate with risk of surgical bleeding in most patients,1 and is no longer widely used or recommended.
The bleeding time (BT) involves making a controlled incision in soft tissue (usually at a site on the forearm) and measuring the time to cessation of bleeding. Anemia and abnormalities of coagulation factors, platelets, or the vasculature may prolong the BT. The BT does not correlate with risk of surgical bleeding in most patients,1 and is no longer widely used or recommended.
![]() The thrombin time (TT) involves the addition of exogenous thrombin to patient plasma, inducing cleavage of fibrinogen to fibrin and the formation of a fibrin clot.
The thrombin time (TT) involves the addition of exogenous thrombin to patient plasma, inducing cleavage of fibrinogen to fibrin and the formation of a fibrin clot.
![]() The most common cause of a prolonged TT is the presence of heparin in the sample, which can be confirmed by documentation of a normalization of the TT when the test is repeated using a heparin-binding agent such as protamine or Heparsorb®.
The most common cause of a prolonged TT is the presence of heparin in the sample, which can be confirmed by documentation of a normalization of the TT when the test is repeated using a heparin-binding agent such as protamine or Heparsorb®.
![]() Abnormalities of fibrinogen and circulating heparin-like anticoagulants also cause a prolonged TT.
Abnormalities of fibrinogen and circulating heparin-like anticoagulants also cause a prolonged TT.
![]() The reptilase time is also used to assess abnormalities of fibrinogen (reptilase cleaves fibrinogen to fibrin). Unlike thrombin, however, reptilase is not inhibited by the presence of heparin. Thus, a prolonged TT in conjunction with a normal reptilase time usually indicates heparin contamination, whereas prolongation of both tests indicates a qualitative abnormality of fibrinogen.
The reptilase time is also used to assess abnormalities of fibrinogen (reptilase cleaves fibrinogen to fibrin). Unlike thrombin, however, reptilase is not inhibited by the presence of heparin. Thus, a prolonged TT in conjunction with a normal reptilase time usually indicates heparin contamination, whereas prolongation of both tests indicates a qualitative abnormality of fibrinogen.
![]() The functional fibrinogen assay assesses fibrinogen concentration by addition of an excess of thrombin to a sample of diluted plasma.
The functional fibrinogen assay assesses fibrinogen concentration by addition of an excess of thrombin to a sample of diluted plasma.
Specialized Coagulation Tests
![]() The anti-Xa assay
The anti-Xa assay
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


