Chapter 17
Disorders of Amino Acid Metabolism, Organic Acidaemias and Urea Cycle Disorders
Marjorie Dixon, Anita MacDonald, Fiona White and Jacky Stafford
Introduction to Inherited Metabolic Disorders
Fiona White
Metabolism
Metabolism describes the chemical processes which occur in the body’s cells to produce energy and other substances needed for normal body functioning. It comprises two elements: anabolism and catabolism. Anabolism is the formation and storage of complex compounds needed for growth, tissue repair and energy storage from simpler molecules. Catabolism is the breakdown of large complex molecules to provide energy for cellular activity and smaller compounds, e.g. amino acids, needed for anabolic reactions or for elimination from the body. Commonly the term metabolism defines the breakdown of food and how its components (carbohydrates, fats and proteins) are transformed into energy via a sequence of chemical reactions (metabolic pathways) which are controlled by large numbers of different enzymes. Enzymes themselves are proteins. These biochemical reactions frequently involve cofactors, often vitamins, which help the specific enzyme function, e.g. vitamin B6 is the cofactor for the enzyme cystathionine β synthase which converts the amino acid homocysteine into cystathionine. Figure 17.1 illustrates the metabolic processes involved in the overall metabolism of carbohydrates, fats and protein including the catabolic processes to produce energy and urea (the product of the detoxification of the nitrogen moiety of amino acids) and anabolic processes to form tissue protein and energy stores, glycogen and lipids.
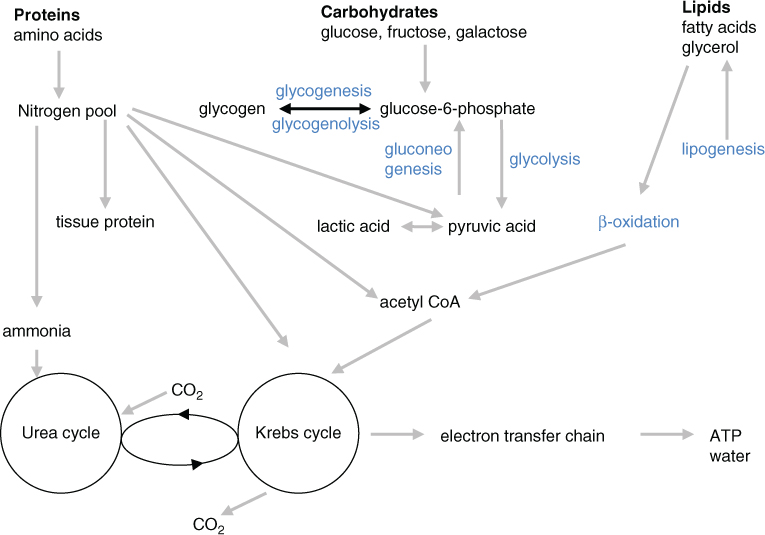
Figure 17.1 Summary of metabolism of carbohydrates, fats and proteins.
Carbohydrate metabolism
Cellular carbohydrate (CHO) metabolism involves both catabolic (glycolysis, glycogenolysis) and anabolic processes (glycogenesis, gluconeogenesis). Carbohydrates, as monosaccharides (glucose, fructose, galactose), are absorbed in the intestine and then transported to the liver where excess glucose, galactose and fructose are converted to glucose-6-phosphate (G-6-PO4). Depending upon energy needs G-6-PO4 undergoes either catabolism to form energy or anabolism to form glycogen, the storage form of glucose, in liver and muscles. To produce energy G-6-PO4 (derived from monosaccharides from dietary CHO or produced from glycogen degradation by glycogenolysis) is converted by a series of enzyme reactions in the glycolytic pathway to form pyruvate or lactic acid, then to acetyl-CoA, which is also produced from fatty acid oxidation and degradation of the carbon skeleton of glucogenic amino acids (Table 17.1). Acetyl-CoA enters the Krebs cycle, also known as the citric acid or tricarboxylic (TCA) cycle, within the mitochondria. Within the Krebs cycle acetyl-CoA, combined with oxaloacetate, undergoes cycles involving eight enzymes, in which reducing equivalents are produced which then enter the electron transfer chain for the production of energy as adenosine triphosphate (ATP). When cells do not require G-6-PO4 for energy production it undergoes glycogenesis to be stored as glycogen until required to restore blood glucose levels. G-6-PO4 can also be produced via pyruvate from protein catabolism of glucogenic amino acids (Table 17.1) or breakdown of glycerol from lipids (gluconeogenesis).
Table 17.1 Classification of amino acids
| Glucogenic only | Ketogenic only | Ketogenic and glucogenic |
| Alanine | Leucine | Isoleucine |
| Arginine | Lysine | Phenylalanine |
| Asparagine | Threonine | |
| Aspartate | Tyrosine | |
| Cystine | Tryptophan | |
| Glutamate | ||
| Glutamine | ||
| Glycine | ||
| Histidine | ||
| Methionine | ||
| Proline | ||
| Serine | ||
| Valine |
Fat metabolism
Dietary fat is present mainly as long chain triglycerides, comprising a glycerol backbone and fatty acids. Dietary fats, and lipids produced endogenously from acetyl-CoA, are initially hydrolysed by lipases into glycerol and free fatty acids. Glycerol is then oxidised to acetyl-CoA via pyruvate. Lipid transport is a complex process and is discussed in Chapter 19. Fatty acids enter the mitochondria via the carnitine transport cycle (medium chain fatty acids enter independently of carnitine) into the β-oxidation spiral in which fatty acids, via a series of enzymes, produce acetyl-CoA and electron carriers. Acetyl-CoA can enter the Krebs cycle or form ketone bodies in the liver. Electron carriers (FADH2 and NAD) enter the electron transfer chain to produce ATP. Acetyl-CoA in excess of requirements for energy production via the Krebs cycle is converted via lipogenesis to stored lipids in adipocytes.
Protein metabolism
Dietary protein is broken down into 20 individual amino acids for absorption. Of these nine are essential or indispensible (histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine) as they cannot be synthesised by the body. The other 11 (alanine, arginine, asparagine, aspartic acid, cystine, glutamic acid, glutamine, glycine, proline, serine, tyrosine) are classified as non essential amino acids (NEAA), or dispensable, as they can be produced from the breakdown of essential amino acids (EAA). A few of the NEAA become conditionally essential in certain disorders or at times of stress. These include arginine, cystine, glutamine, glycine, proline and tyrosine and in such circumstances must be provided in the diet.
Amino acids enter the body’s nitrogen pool (Fig. 17.2) and are used to form tissue protein for growth and body tissue repair, or are catabolised by two processes. The amine group (nitrogen moiety) undergoes transamination, in which it is transferred to a keto acid producing a keto acid and an amino acid, and then deamination to produce ammonia which is detoxified via the urea cycle to produce urea, which is then excreted in urine. The carbon skeletons of amino acids (organic acids) are glucogenic, ketogenic or both (Table 17.1).
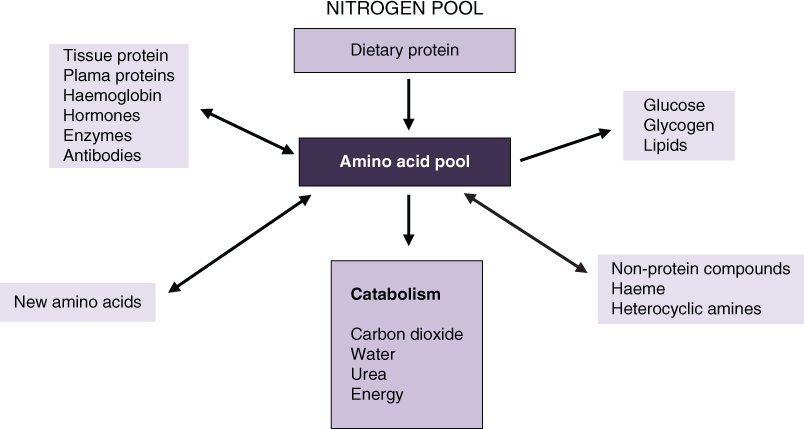
Figure 17.2 Function of the amino acid pool.
Glucogenic amino acids degrade
- directly to Krebs cycle intermediates
- via acetyl-CoA or acetoacetyl-CoA to oxaloactetate which is either used in the Krebs cycle or can be converted to phosphoenolpyruvate and used to produce G-6-PO4 via gluconeogenesis
- to pyruvate which can either be used to produce G-6-PO4 via gluconeogenesis or converted irreversibly to acetyl-CoA by the pyruvate dehydrogenase (PDH) complex. Acetyl-CoA can then enter the Krebs cycle
Ketogenic amino acids degrade to acetoacetate or acetyl-CoA and are used in fatty acid synthesis or ketone body production (Fig. 17.3).
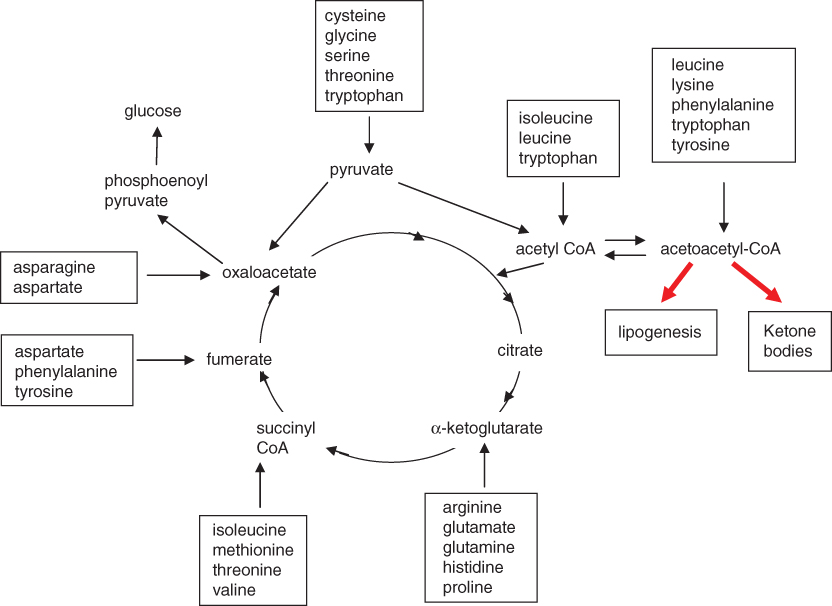
Figure 17.3 Catabolism of amino acids.
Control and regulation of metabolism
To maintain homeostasis within cells metabolic pathways have to be finely balanced (Fig. 17.4). This is achieved by either
- flux through pathways, increasing or decreasing by intracellular self-regulation of reactions (feedback) by responding to concentrations of substrates or products, e.g. a decreased concentration of an essential product causes enzyme activity in the metabolic pathway to increase and produce more product from the substrate
- responding to signals from other cells, involving hormones and growth factors
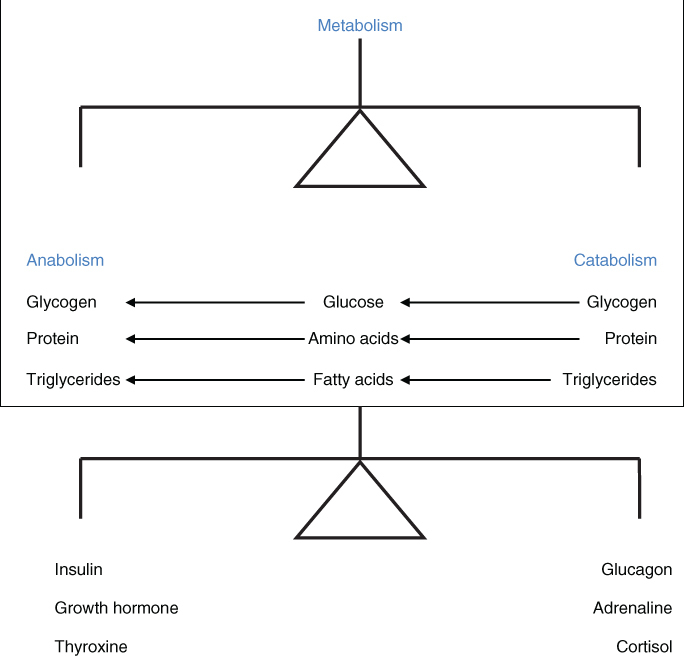
Figure 17.4 Regulation of metabolic balance.
Influence of hormone regulation
Hormones are released according to the metabolic status including whether in the fed or starved state and stress. Their regulation of metabolic pathways play a crucial role by
- altering the availability of substrates, e.g. sympathetic response during exercise increasing the mobilisation of fatty acids, insulin being released in response to high blood glucose levels and glucagon being released when glucose levels are low
- influencing enzyme activity, e.g. epinephrine, a fight and flight hormone, increases phosphofructokinase (PFK) activity, an important enzyme in glycolysis. In addition glucocorticoids and thyroid hormones also affect energy metabolism
Table 17.2 summarises the action of hormones on metabolic pathways.
Table 17.2 Summary of the action of hormones on metabolic pathways
| Hormone | Anabolic/ catabolic | Stimulates | Inhibits | Blood levels | ||
| Glucose | Fatty acids | Amino acids | ||||
| Insulin released in response to
| Anabolic | Uptake of glucose by muscle and adipose tissue
Cellular uptake of amino acids
|
|  |  |  |
| Glucagon released in response to
| Catabolic | Glycogen breakdown and formation of glucose
Fat breakdown
|
|  |  |  |
| Epinephrine released during exercise and stress (stimulation of sympathetic nervous system) | Catabolic | Stimulates glucagon and cortisol release Glycogen breakdown and formation of glucose
Fat breakdown
|
|  |  |  |
| Cortisol Released due to physiological and emotional stress Supports effects of decreased insulin and increased glucagon secretion | Catabolic | Decreased glucose, fatty acid and amino acid uptake by all tissues except brain
|  |  |  | |
| Growth hormone Released in response to
| Anabolic | Muscle protein synthesis Mobilisation of free fatty acids
Reduced cellular glucose uptake
| Glucose uptake by muscles (glucose sparing) |  |  |  |
| Thyroxine Slow response to metabolic changes Increases sensitivity to epinephrine, glucagon and growth hormone Increases basal metabolic rate | Catabolic | Increases ATP production
| indirectly | |||
Inherited metabolic disorders
Inherited metabolic disorders (IMD) are genetically inherited biochemical disorders of specific enzymes or proteins causing a block in a normal metabolic process of protein, CHO or fat metabolism. As a result there is an abnormality in the normal metabolic process and accumulation of intermediary metabolites which cannot be further metabolised. This concept of IMD was originally conceived by Garrod [1] in 1902 and in 1908 he went on to describe such disorders including alkaptonuria, albinism, pentosuria and cystinuria [2] which are all inherited in an autosomal recessive trait. IMD can be categorised in a number of ways. Based on their pathophysiology they can be considered in three distinct groups.
- Disorders of intermediary metabolism leading to intoxication
These include disorders in which the enzyme deficiency leads to accumulation of intermediary metabolites proximal to the block (B) and the formation of alternative products (D) producing acute or chronic toxic effects which result in disease manifestations and may also lead to deficiencies of products downstream of the block (C).
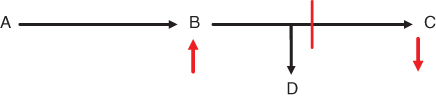
Examples of such disorders include the amino acidopathies (phenylketonuria, maple syrup urine disease, homocystinuria, tyrosinaemias); organic acidaemias; urea cycle disorders; disorders of sugar intolerance (galactosaemia, hereditary fructose intolerance); metal intoxication disorders (Menkes disease, Wilson disease) and porphyrias. These disorders all have a symptom free period after birth and then present with acute (either in the neonatal period or a later onset) or chronic signs of ‘intoxication’. Acute symptoms can be triggered by dietary intake, catabolism, fever, intercurrent illness. Chronic, progressive presentations include developmental delay, failure to thrive, cardiomyopathy, hepatomegaly, lens dislocation.
- Disorders of intermediary metabolism involving energy metabolism
These include disorders in which the enzyme deficiency results in lack of energy production or energy utilisation within organs including liver, muscle, brain, myocardium. These disorders can be subdivided into defects of mitochondrial or cytoplasmic energy:
- mitochondrial energy disorders include disorders of fatty acid oxidation, ketone body defects, congenital lactic acidaemias, respiratory chain disorders
- cytoplasmic energy disorders include disorders of glycolysis, glycogen metabolism, gluconeogenesis, glucose transport and hyperinsulinism and disorders of creatine metabolism
Symptoms may include hypoglycaemia, lactic acidosis, hepatomegaly, cardiomyopathy, myopathy, hypertonia, failure to thrive, sudden infant death.
- mitochondrial energy disorders include disorders of fatty acid oxidation, ketone body defects, congenital lactic acidaemias, respiratory chain disorders
- Disorders involving complex large molecules
These are disorders in which complex molecules are incompletely catabolised and accumulate in organelles resulting in varied symptoms including progressive loss of function of affected body systems, organomegaly and dysmorphic features. They include disorders of lipoprotein and cholesterol metabolism, lysosomal storage disorders, peroxisomal disorders, disorders of glycosylation, disorders of sterol synthesis and disorders of purine and pyrimidine metabolism.
Management
A significant number of IMD are treatable, often by therapeutic dietary intervention. The main forms of nutritional therapy in IMD are:
- substrate reduction in which the metabolite which accumulates, due to the enzyme deficiency, is restricted (if it is an essential nutrient) or as far as possible removed from the diet. Such a treatment strategy is employed in managing
- disorders of amino acid metabolism
- disorders of galactose and fructose metabolism
- lipoprotein disorders
- disorders of fatty acid oxidation
- some peroxisomal disorders, e.g. Refsum’s disease
- disorders of amino acid metabolism
- provision of products normally produced downstream of the block and which, as a result of the block, are conditionally essential to prevent deficiency
- tyrosine supplementation in phenylketonuria
- arginine in urea cycle disorders
- maintaining normoglycaemia by regular feeding and/or CHO supplements in
- glycogen storage disorders
- disorders of fatty acid oxidation
- glycogen storage disorders
- cholesterol in Smith–Lemli–Opitz syndrome
- mannose in CDG 1b (phosphomannose isomerase deficiency)
- tyrosine supplementation in phenylketonuria
- stimulation of residual enzyme activity by use of pharmacological doses of cofactors, often vitamins
- vitamin B12 in methylmalonic acidaemia
- pyridoxine (vitamin B6) in homocystinuria
- riboflavin (vitamin B2) in multiple acyl-CoA dehydrogenase deficiency
- sapropterin in some forms of biopterin responsive phenylketonuria
- vitamin B12 in methylmalonic acidaemia
- providing alternative substrates
- use of glucose and medium chain triglycerides in long chain fat oxidation disorders
- use of the ketogenic diet in Glut 1 deficiency syndrome to produce ketones as an alternative energy source to glucose by the brain
- use of glucose and medium chain triglycerides in long chain fat oxidation disorders
In many IMD the promotion of anabolism and avoidance of catabolism is vital to prevent metabolic decompensation occurring. Where acute metabolic decompensation occurs with the risk of further irreversible damage, particularly neurological, the use of emergency dietary regimens is imperative. Detailed discussion of dietary treatment approaches are given in the relevant sections of this and the following chapters.
Patients with IMD are invariably complex to treat and it is important they are managed by an experienced metabolic team including doctors, specialist dietitians, clinical nurse specialists, biochemists, pharmacists and psychologists in a metabolic centre. Local professionals and hospital teams also form an integral part of the care for metabolic patients.
Newborn screening
Early detection is important to ensure a good outcome in many IMD, starting treatment before significant or any damage is done. Newborn screening (NBS) aims to detect affected infants before symptoms occur to allow early implementation of treatment. Phenylketonuria was the first IMD to be part of NBS (from the end of the 1960s); screening has been expanded to include a variety of other disorders, depending on local health policy. Further information is given in disorder specific sections.
Inheritance
The concept of IMD being due to a block in a metabolic process, with the accumulation of intermediary metabolites leading to disease, was introduced by Garrod [1, 2]. In 1911 Johannsen [3] introduced the theory that genes were responsible for inheritance and in 1941 Beadle and Tatum [4] hypothesised the one gene, one enzyme theory. The molecular disease concept in which gene mutations alter protein structure by changing a single amino acid within the protein was introduced by Pauling et al. [5] and Ingram [6] with their work on sickle cell anaemia.
The structure of DNA was described by Watson and Crick in 1953. DNA contains information as a code made up of four different chemical bases. The sequence of bases provides information for building and maintaining body structures. DNA sequences are made up of exons and introns. Exons are coding sequences which contain information to encode for a protein. Introns, which occur between exons, do not code for proteins. The process of a gene expressing for a protein involves two significant steps, transcription and translation. Transcription, within the cell nucleus, involves the transfer of information in the DNA to messenger RNA (mRNA). mRNA carries this information out of the nucleus into the cytoplasm for translation where ribosomes read the sequence of mRNA bases. Three base (codon) sequences code for specific amino acids (AA). Transfer RNA (tRNA) then assembles the protein by sequentially adding AA until a stop codon (a three base sequence which does not code for an AA) is reached [7].
Genetic disorders arise due to mutations (change) in the DNA sequence. An error in the coding sequence of a gene can result in no protein or an abnormal protein or enzyme being produced. Many mutations are benign (silent) whereas others lead to variable severity of defect. Mutation types include point mutations, missense mutations, nonsense mutations, deletions, insertions, inversions, duplication, frameshift mutations (Table 17.3). Further information can be obtained at www.genetichealth.com and www.ghr.nlm.nih.gov/handbook/mutationsanddisorders/possiblemutations.
Table 17.3 Types of mutations
| Mutation type | Change in amino acid (AA) code | Effect |
| Point | Single base pair change | Benign unless causes change in AA code |
| Missense | Single base pair change | Substitution of one AA for another in the protein chain. Can have serious consequences |
| Nonsense | Single base pair change or premature termination of AA code (stop codon) | Signals cell to stop building a protein, thus producing a shortened protein which may function improperly or not at all |
| Insertion | Adds a piece of DNA which changes number of DNA bases | Protein product produced may not function correctly |
| Deletion | Removes piece of DNA | Function of resulting protein may be altered |
| Duplication | Piece of DNA abnormally copied once or multiple times | Resulting protein function may be altered from normal |
| Frameshift | Loss or addition of one or more DNA bases causing change in grouping of the triplet base pair coding for AA Deletions, insertions and duplications can also result in frameshift mutations | Usually results in non functioning protein |
| Splice site mutations | Introns (bulk of the DNA within a gene which is not translated) not spliced from mRNA | Incorrect protein is produced |
Gene mutations result in a genotype which predicts residual enzyme activity. Single genes do not function independently but interact with other genes and the individual’s environment resulting in the disease phenotype. There is not always an exact genotype–phenotype correlation.
Patterns of inheritance
Autosomal recessive (AR) inheritance
In AR inheritance faulty (mutated) copies of the gene have to be inherited for the disease to be expressed (Fig. 17.5). Parents are obligate heterozygotes (carriers) but do not have the disease. They may carry the same or a different gene fault (mutation). Affected offspring are either homozygous with two copies of the same gene fault (mutation) or compound heterozygotes with two different gene faults (mutations). There is a 25% (one in four) risk of having an affected child with each pregnancy. Unaffected children have a 66% (two out of three) chance of being a carrier. Many IMD are inherited in this way including phenylketonuria (PKU), maple syrup urine disease (MSUD), galactosaemia, glycogen storage disease (GSD) Ia, medium chain acyl CoA dehydrogenase deficiency (MCADD).
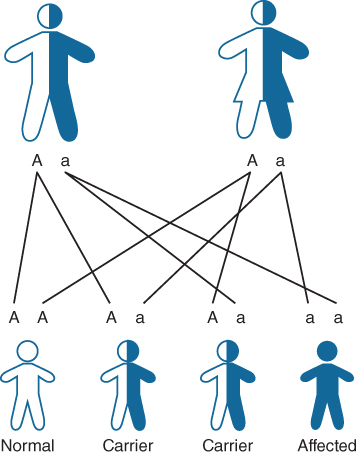
Figure 17.5 Autosomal recessive inheritance (http://www.geneticseducation.nhs.uk).
When individuals affected with an AR disorder have children they will all inherit the faulty (mutated) copy of the gene. The risk of their children actually having the same AR disorder depends upon whether their partner is affected with the same AR disorder or carries a faulty (mutated) copy of the same gene. Children of parents who both have the same AR disorder will all inherit the disorder. The risk of offspring inheriting the disorder when one parent has an AR disorder depends upon the carrier frequency rate for the disorder. For example PKU has a carrier frequency of approximately 1 in 50 which gives a 1 in 100 (1%) chance of any child of a parent with PKU inheriting the condition.
Autosomal dominant (AD) inheritance
AD inheritance only requires one copy of a faulty (mutated) gene to inherit the disorder; this can be inherited from either parent (Fig. 17.6). There is a 50% risk of occurrence with each pregnancy. Unaffected offspring will have two normal copies of the gene. Dominant mutations can occur sporadically (de novo) with both parents having no mutations. This is often seen in Glut 1 deficiency syndrome. Occasionally an AD disorder can occur as the result of a germ line mutation; parents usually have a normal phenotype but one of the parents has a mutation occur in some of his/her sperm/eggs. AD conditions can occur in a homozygous state in which case they can be very severe or lethal. Familial hypercholesterolaemia (FH) is an autosomal dominant disorder; heterozygotes are symptomatic but homozygotes are more severe, often dying from fatal myocardial infarction in early adult life. Individuals with heterozygous AD disorders (only one copy of the faulty gene) have a 50% chance of passing on the faulty gene. All children to individuals with homozygous AD disorders will inherit the disorder.
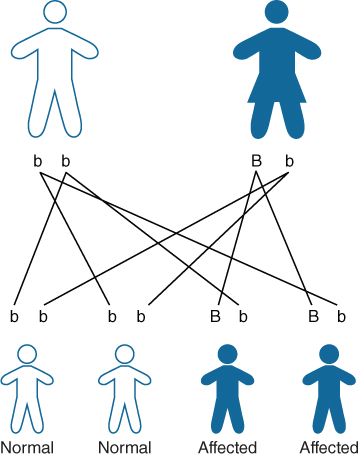
Figure 17.6 Autosomal dominant inheritance (http://www.geneticseducation.nhs.uk).
X-linked inheritance
Mutations occur in genes on the X chromosome (Fig. 17.7). X-linked recessive single gene defects affect males as they only have one X chromosome and if the gene fault/mutation is passed on from a carrier mother then they are affected. Females carry the gene fault/mutation on one of their two X chromosomes. Depending upon the degree of X inactivation (how many of the mutant X chromosomes are active) females can be symptomatic to a greater or lesser extent, e.g. in ornithine transcarbomylase (OTC) deficiency some carrier females present with hyperammonaemia. Males with X-linked disorders will pass on the faulty gene to all of their daughters; sons are unaffected. Examples of X-linked IMD include adreoleukodystrophy, OTC deficiency, X-linked GSD type IX.
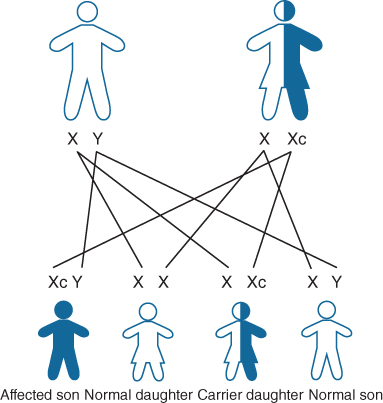
Figure 17.7 X-linked inheritance (http://www.geneticseducation.nhs.uk).
Mitochondrial inheritance
Mitochondria contain their own DNA, which originates in the egg and encodes for enzymes involved in electron transport and energy metabolism. Some mitochondrial encoded polypeptides join with nuclear originated protein to form a complete enzyme. Thus mitochondrial disorders can be inherited in maternal, recessive, X-linked or dominant fashion dependent upon the defective polypeptide.
Cells contain multiple mitochondria and therefore many alleles for one gene. When some alleles are mutated the cells contain a mixture of normal and mutated alleles, known as heteroplasmy. Depending on the number of mutated mitochondria in the cell or tissue, diseases with the same mutation can be expressed differently. Examples of mitochondrial inherited IMD include Leigh syndrome, Pearson syndrome, mitochondrial encephalomyopathy lactic acidosis and stroke like episodes (MELAS).
Summary
There are over 500 different IMD currently identified. A significant number are treatable, often by therapeutic dietary intervention as the sole treatment, or in combination with pharmacological agents. These are discussed further in the following sections.
Amino Acid Disorders
Anita MacDonald Fiona White
In this section the following amino acid disorders are considered: phenylketonuria (PKU) and tetrahydrobiopterin (BH4) deficiencies, maple syrup urine disease (MSUD), homocystinuria (HCU) and tyrosinaemia. For each condition, the prevalence, biochemistry, genetics, pathophysiology, diagnosis, outcome and non dietetic treatment is described separately. All these amino acid conditions share the same principles of dietary management. Therefore, the dietary management for all conditions is considered in one section, with any specific feature for each condition being individually discussed.
The following definitions are used throughout this chapter:
- substrate/precursor amino acid: an amino acid that cannot be catabolised due to an enzyme defect in a metabolic pathway, e.g. phenylalanine in phenylketonuria; methionine in homocystinuria.
- L-amino acid: an amino acid, with the exception of glycine, that can be found in two stereoisomeric forms (L and D forms). Only L-amino acids are used by cells. D-amino acids are not naturally found in proteins but are important in the structure of bacteria.
- protein equivalent: the amount of protein provided by an L-amino acid supplement. Total protein equivalent refers to protein sourced from an L-amino acid supplement and natural protein from the diet.
- tandem mass spectrometry (MS/MS): the analysis technology commonly used in newborn screening. It consists of a specialised instrument that detects molecules by measuring their weight or mass electronically and results are displayed (graphically) in the form of a mass spectrum.
Phenylketonuria
PKU is an autosomal recessive inborn error of amino acid metabolism, caused by a deficiency of the hepatic enzyme phenylalanine hydroxylase (PAH) (phenylalanine 4-mono-oxygenase, EC 1.14.16.1), which catalyses the hydroxylation of phenylalanine to tyrosine, the rate limiting step in phenylalanine catabolism. The enzyme deficiency leads to increased phenylketones (hence phenylketonuria); accumulation of phenylalanine resulting in hyperphenylalaninaemia; low tyrosine concentrations; lower dopamine, norepinephrine and serotonin production [8].
This high level of brain phenylalanine is possibly the chief cause of neurotoxicity [9]. It is thought to interfere with cerebral protein synthesis [10], increases myelin turnover [8] and inhibits neurotransmitter synthesis [11], the latter caused by impaired uptake of non phenylalanine large neutral amino acids in the presence of elevated plasma phenylalanine concentrations. Without treatment most children develop profound and irreversible intellectual disability [12]. Fortunately in the developed world symptomatic PKU is rarely seen due to the introduction of newborn screening and early treatment, but newborn screening is still uncommon in some regions of the Middle East, Asia and Africa.
The prevalence of PKU is given in Table 17.4. The global prevalence in screened populations is approximately 1 in 12 000 with an estimated carrier frequency of 1 in 55 [13]. There is an equal gender ratio between boys and girls.
Table 17.4 Prevalence of PKU
| Population | Prevalence |
| Turkish [14] | 1 in 4000 |
| Irish [15] | 1 in 6200 |
| Eastern Europe [15] | 1 in 3000 to 1 in 33 000 |
| Arabic population [16] | Up to 1 in 6000 although variable between Arab countries |
| China [17, 18] | 1 in 10 000 to 1 in 17 000 |
| Western Europe [15] | 1 in 7000 to 1 in 33 000 |
| West Midlands (UK) [19] | 1 in 12 600 northwest Europeans 1 in 14 500 Pakistani population |
| Southern Europe [15] | 1 in 4000 to 1 in 36 000 |
| Poland [15] | 1 in 8000 |
| Russia [15] | 1 in 8000 |
| USA [20] | 1 in 15 000 |
| Finnish [21] | 1 in 100 000 |
| African [22] | 1 in 100 000 |
| Japanese [23] | 1 in 70 000 |
Countries use different upper ‘cut-off’ phenylalanine concentrations to diagnose PKU.
Classification of PKU
There is a broad continuum of phenotypes in PKU ranging from severe to milder forms. The need for diet therapy and the amount of dietary phenylalanine tolerated gives an indication of severity [24]. PKU is generally classified by the severity of hyperphenylalaninaemia at diagnosis.
- Classical or severe PKU is usually characterised by blood phenylalanine concentrations >1200 µmol/L, with a phenylalanine tolerance no greater than 250 mg/day to maintain plasma phenylalanine concentrations <360 µmol/L [25].
- Moderate PKU is classified with diagnostic phenylalanine concentrations of 600–1200 µmol/L. Most patients with moderate/severe PKU tolerate between 200 and 700 mg/day phenylalanine whilst maintaining plasma phenylalanine concentrations <360 µmol/L.
- Patients with persistent hyperphenylalaninaemia, or mild PKU, present with plasma phenylalanine concentrations between 120 and 600 µmol/L on a normal diet. If phenylalanine concentrations are maintained within these limits without treatment, there is little evidence to suggest patients are at risk of intellectual, neurological or neuropsychological impairment [26]. Some centres, however, advocate treatment when phenylalanine concentrations are >360 µmol/L [25, 27, 28].
- Moderate PKU is classified with diagnostic phenylalanine concentrations of 600–1200 µmol/L. Most patients with moderate/severe PKU tolerate between 200 and 700 mg/day phenylalanine whilst maintaining plasma phenylalanine concentrations <360 µmol/L.
Tetrahydrobiopterin deficiencies
About 1%–2% of hyperphenylalaninaemia is caused by mutations in genes coding for enzymes involved in tetrahydrobiopterin (BH4) cofactor biosynthesis or regeneration [29], affecting phenylalanine homeostasis and catecholamine and serotonin biosynthesis [30]. The BH4 deficiencies are treated with neurotransmitters with or without BH4 supplementation (Table 17.5). A low phenylalanine diet may be prescribed for hyperphenylalaninaemia. Without treatment [except pterin-4-α-carbinolamine dehydratase (PCD) deficiency] the disorders are generally characterised by progressive developmental delay, irritability, motor dysfunction, impaired muscle tone, movement disorders and seizures [13, 31]. Symptoms may be subtle only in the newborn period [13].
Table 17.5 Tetrahydrobiopterin deficiencies with raised phenylalanine concentrations: treatment and use of low phenylalanine diet [31]
| Condition | Mean (range) plasma phenylalanine (µmol/L) before onset of treatment | Treatment | Use of low phenylalanine (phe) diet |
| GTP cyclohydrolase deficiency (GTPCH deficiency) | 533 (55–2930) | L-dopa/carbidopa, 5-hydroxytryptophan, BH4 | ≤5% given low phe diet |
| 6-pyruvoyl-tetrahydropterin synthase deficiency (PTPS deficiency) | Severe: 829 (121–2251) Mild: 1111 (41–3805) | L-dopa/carbidopa, 5-hydroxytryptophan, BH4 | 15% given low phe diet |
| Dihydropteridine reductase deficiency (DHPR deficiency) | 717 (22–3207) | L-dopa/carbidopa, 5-hydroxytryptophan, folinic acid | Many given low phe diet* |
| Pterin-4-alpha-carbinolamine dehydratase deficiency (PCD deficiency) | 533 (183–1850) | Transient and benign without neurotransmitter deficiency or neurological symptoms | May be treated with BH4/low phe diet |
* In DHPR deficiency BH4 supplementation may lead to an increased risk of central nervous system folate deficiency [13].
Biochemistry
Phenylalanine hydroxylase catalyses the hydroxylation of phenylalanine to tyrosine in the presence of the cofactor BH4 and several enzymes that serve to regenerate BH4, e.g. dihydropteridine reductase and pterin-4-α-carbinolamine [32] (Fig. 17.8).
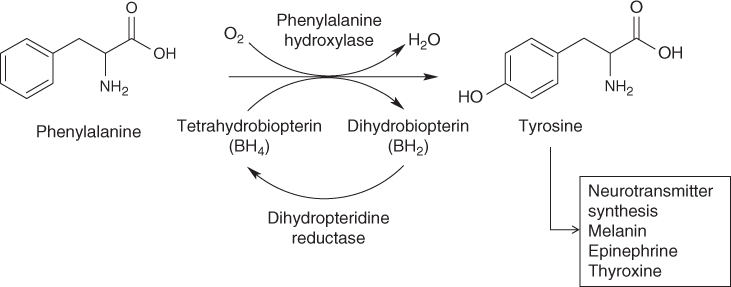
Figure 17.8 Hydroxylation of phenylalanine.
Normally hydroxylation of phenylalanine contributes up to 50% of the tyrosine that is incorporated into tissue protein [33]. In PKU there is an inability to convert phenylalanine into tyrosine. Subsequently low to normal or reduced concentrations of tyrosine are common in untreated patients. In addition the phenylalanine : tyrosine ratio, normally around 1:1, is significantly increased. Tyrosine is essential for the synthesis of the catecholamine neurotransmitters such as dopamine and norepinephrine.
Genetics
PKU is caused by mutations in the gene encoding the phenylalanine hydroxylase enzyme (PAH). The gene is on the long arm of chromosome 12 in the band region of q22-q24.1 [34]. There are more than 800 disorder causing mutations in the PAH gene [35, 36]. Different mutations affect the activity of PAH dissimilarly ranging from little or no effect to complete suppression of the activity of the enzyme [24]. There is a good correlation between genotype and phenotype [37]. Consequently genotypes with more severe mutations will result in a moderate to classical phenotype with higher untreated blood phenylalanine concentrations. Mild mutations in PAH will result in a phenotypically less severe form of PKU. In those cases where a patient is heterozygous for two mutations of PAH (i.e. each copy of the gene has a different mutation) the milder mutation will predominate. Spain has an especially high prevalence of mild PKU [25].
There are a small number of patients with so called severe PKU mutations who have escaped intellectual disability despite high blood phenylalanine concentrations and poor dietary control. These patients appear to have near normal brain phenylalanine concentrations despite high blood phenylalanine levels [38].
Diagnosis
In the UK PKU is detected on routine neonatal screening by collecting blood spots from the heel onto filter paper between days 5 and 8 of life, which are then analysed by tandem mass spectrometry (MS/MS) [39] (see www.newbornscreening-bloodspot.org.uk for screening guidelines for PKU in the UK). Infants with a phenylalanine level >240 µmol/L should be referred to a designated PKU team and assessed within 24 hours of a positive screening result. A repeat blood sample to recheck phenylalanine and tyrosine measurements, together with blood for dihydropteridine reductase and blood samples for pterins (neopterin and biopterin) to diagnose tetrahydrobiopterin deficiencies, should be taken. A low phenylalanine diet should be commenced immediately providing blood phenylalanine concentrations are ≥400 µmol/L (although some centres commence treatment when blood phenylalanine concentrations are ≥600 µmol/L [27]). It is a UK standard that all infants have commenced dietary treatment by 14 days of age [39].
It is good practice to test any new sibling of a proband case with PKU at 48–72 hours and then repeat at the usual newborn screening time (days 5–8). In the UK DNA analysis is used mainly for prenatal testing only or to detect carrier status of an ‘at risk relative’ [38].
Treatment guidelines
Diet is the basis of treatment for patients with PKU with blood phenylalanine concentrations consistently ≥400 µmol/L. It consists of a limited and controlled amount of natural protein derived from food sources to provide essential phenylalanine requirements, with the majority of nutrient requirements being met by a phenylalanine free source of L-amino acids, usually supplemented with added micronutrients (a protein substitute). The overall aim of treatment should be to ‘achieve an optimal cognitive and psychological development and well being of the child’ [40]. Published guidelines have been unanimous in their requirement for neonatal screening and strict blood phenylalanine control in the first 10 years of life [20, 41–43], but there has been little agreement about the upper blood phenylalanine threshold to commence treatment, phenylalanine target concentrations for adolescents and adults, the need for long term treatment, frequency of blood phenylalanine monitoring and use of alternative treatments such as large neutral amino acids (LNAA) and sapropterin. Currently a group of health professionals, working with the European Society for PKU (ESPKU), are developing PKU guidelines which, it is hoped, will be adopted throughout Europe and beyond. Recommended blood phenylalanine concentrations are given in Table 17.6.
Table 17.6 Recommended blood phenylalanine concentrations throughout life
| Target blood phenylalanine concentrations (µmol/L) | ||||
| Age | UK [43] | Germany [42] | USA [20] | France [40] |
| 0 months to 6 years | 120–360* | 40–240 | 120–360 | 120–300 |
| At school age (4–5 years) upper phenylalanine level is increased to 480 µmol/L | ||||
| 7–9 years | 120–480 | 40–240 | 120–360 | 120–300 |
| 10–12 years | 120–700 | 40–900 | 120–360 | <900 |
| 13–15 years | 120–700 | 40–900 | 120–600 | <900 |
| 16–18 years | 120–700 | 40–1200 | 120–900 | <900 |
| >18 years | 120–700 | 40–1200 | 120–900 | <1500 |
Normal physiological plasma phenylalanine concentration in infants, children and adolescents is approximately 60 µmol/L [8].
Recent meta-analysis of blood phenylalanine concentrations and intelligence quotient (IQ) [44] support the approach that treatment should be continued in adulthood. Furthermore, recent meta-analysis of data from reaction times studies suggested that upper phenylalanine concentrations should be 320 µmol/L for children aged 7–13 years; 570 µmol/L for adolescents aged 14–18 years [45].
Outcomes in early diet treated PKU
Neurocognitive and neuropsychological
If untreated, PKU leads to global intellectual disability [46], significant delays in developmental milestones, hyperactive behaviour with autistic features, seizures, eczema, musty body odour and light pigmentation (eyes, hair and skin). If treatment is started within the first 3 weeks of life, irreversible intellectual disability is prevented. Most early treated children who have commenced diet by 4 weeks of age fall within the broad normal range of general ability, attain expected educational standards and lead independent lives as adults [25]. Outcome, however, is dependent on the quality of blood phenylalanine control [47] and children may have subtle defects in IQ, attention, processing speed, fine motor skills and perception and visual-spatial abilities [48]. Executive function (working memory, planning, organisation and inhibitory control) may be impaired [49, 50] (Table 17.7). Sustained attention and reaction time is reduced [51]. In adolescents meta-analysis indicates that any relaxation of blood phenylalanine concentrations >600 µmol/L is associated with slower processing speed [45]. Psychological and psychiatric disturbances may develop including anxiety related disorders (phobias/panic attacks), low level depression, attention deficit/hyperactivity, low mood and agoraphobia [52]. Associations between the quality of blood phenylalanine control and behaviour problems, sustained attention and lower IQ are well documented [52, 53].
Table 17.7 Neurocognitive and neuropsychological outcome reported in PKU
| Suboptimal neurocognitive/neuropsychological outcome | Authors |
| Effect on IQ A meta-analysis of 40 studies indicated that during the critical periods (0–12 years of age) every increase of 100 µmol/L of phenylalanine over target was associated with a 1.3–3.1 point reduction in IQ. Similar significant correlations were observed between lifetime IQ and mean lifetime phenylalanine level for early treated patients where each 100 µmol/L increase in phenylalanine predicated a 1.9–4.1 reduction in IQ | [47] |
| A meta-analysis examining neuropsychological and intellectual presentations in continuously treated adolescents and adults (113 patients with PKU compared with 107 controls) found a substantially lower average IQ than the control group. They also differed from control adults on processing speed, attention, inhibition and motor control | [54] |
| A regression model of the relationship between IQ, lifetime blood phenylalanine concentrations and blood phenylalanine variability from 46 children aged 2–15 years showed that IQ decreased by 4.3 points for every 1 point increase in standard deviation (SD) of blood phenylalanine | [55] |
| Tremor, deficits in fine motor abilities: hand–wrist steadiness, finger–hand dexterity and hand–wrist speed | [56] |
| Deficits in motor speed and motor control | [57–59] |
| Deficits in working memory | [60, 61] |
| Reduced reaction times | [51, 62] |
| Deficits in sustained attention | [63] |
| Information processing slower and less efficient in children and adults with PKU | [45, 58, 62, 64–66] |
| Children and adolescents with PKU showed lower performance in several executive functioning skills including initiation of problem solving, concept formation and reasoning | [49, 50, 66, 67] |
| Autism, attention deficit hyperactivity | [68] |
| Psychiatric emotional and behavioural characteristics in children Attention problems, school problems, less achievement motivation, decreased social competence, decreased autonomy and low self-esteem | [67, 69] |
| Psychiatric emotional and behavioural characteristics in teenagers/adults Depressed mood, generalised anxiety, phobias, decreased positive emotions, low self-esteem, social maturity deficits, social isolation or withdrawal, delayed autonomy, lower rate of forming normal adult relationships | [52, 63, 67, 69–73] |
Quality of life
Health related quality of life in PKU is generally comparable to a healthy population [63, 73–75]. In a group of 172 adults with PKU 76% were in full-time employment, 23% had obtained university degrees and 67% remained single [76]. In a further study more adults with PKU lived with their parents with fewer having children compared with controls [73]. Increasing non adherence in adolescents is associated with better quality of life [77] although parents worry more about school performance when their child’s blood phenylalanine concentration results are poor [78]. In direct contrast returning to a low phenylalanine diet in adults is associated with improved quality of life [79, 80].
Growth and nutrition
Growth
There are few differences in growth [81, 82] and body composition [83] between children with PKU and the general population. In the 1990s there were studies describing low length for age in infants and young children [84–86], but height for age improved with time [85].
Overweight
Early studies indicated that PKU children may have a higher prevalence of overweight [87–89]. Although the rate of overweight [81, 90, 91] and percentage of body fat is high in older girls, particularly over the age of 11 years, many researchers have demonstrated it is comparable to the general population [92–94]. Factors associated with overweight in PKU include relaxation of strict diet therapy, a high carbohydrate intake and low activity levels [93].
Bone health
Low bone mineral density (BMD) in children [95] and osteopenia in adults has been reported [96, 97]. There is some suggestion of increased spontaneous osteoclastogenesis [98], consistent with increased bone resorption markers, in affected patients [96, 98]. It is unclear if the deficits in bone mineralisation relate to the disorder or its treatment. Possible reasons for poor BMD include amino acid imbalances; inadequate intake of natural protein, calcium, phosphorus, magnesium [99], vitamin D [96] or n-3 polyunsaturated fatty acids [100]; low calcium/protein ratios; poor dietary adherence with micronutrient supplemented L-amino acids [101, 102]; low plasma magnesium, zinc and copper concentrations [99]; elevated plasma phenylalanine concentrations [103]; lack of weight bearing exercise [95].
Vitamin B12 deficiency
Vitamin B12 deficiency is mainly reported in adolescents or adult patients who have either relaxed their diets and are less adherent with phenylalanine free L-amino acid supplements; or stopped treatment but are not eating a normal protein intake particularly animal protein which is the major source of vitamin B12 [104–107]. Symptoms such as spastic paraparesis, tremor and slurred speech are associated with deficiency, but patients may be unaware of the early manifestations of vitamin B12 deficiency as they appear insidiously [105].
Minerals and trace minerals
In the early years of PKU treatment biochemical selenium deficiency was common [108–110]. Selenium is now added to the majority of phenylalanine free L-amino acid supplements, thereby improving selenium status [111]. Low plasma zinc concentrations are still described [112–114], despite generous zinc supplementation in phenylalanine free L-amino acid supplements. Iron deficiency anaemia [115, 116], low haemoglobin [117] and ferritin [117, 118] concentrations are documented in children on diet therapy.
Antioxidants
Altered antioxidant capacity is observed in PKU both in human and animal models [119, 120] and markers of lipid, protein and DNA damage have been reported. Although this may be associated with an increased free radical generation, it may also be due to depletion of the antioxidant micronutrients [121] or a combination of both [122].
Other treatment options
A number of new treatment options, either as an adjunct therapy to a low phenylalanine diet or to replace diet, are available or in development.
Large neutral amino acids
Large neutral amino acids (LNAA)—histidine, isoleucine, methionine, phenylalanine, threonine, tryptophan, tyrosine, valine—are carried into the brain via the L-type amino acid carrier. They compete for the carrier and their rate of entry into the brain depends upon plasma concentrations. High concentrations of plasma phenylalanine will reduce the entry of the other LNAA into the brain and result in higher brain phenylalanine levels. In PKU, giving large amounts of phenylalanine free LNAA competes with phenylalanine and may reduce its influx into the brain [123, 124]. It may increase both brain neurotransmitter concentrations and brain essential amino acid concentrations [125]. It is suggested that LNAA may have a secondary effect by decreasing blood phenylalanine concentration and inhibiting phenylalanine entry into the circulation [126]. Transport of LNAA across membranes (from gut to blood and from blood to brain) is facilitated by the transporter proteins, which selectively bind to the LNAA. Clinical studies are summarised in Table 17.8.
Table 17.8 Studies examining large neutral amino acids (LNAA) in PKU
| Study detail | Authors |
| [127] |
| [128] |
| [129] |
| [130] |
| [131] |
| [132] |
The use of phenylalanine free LNAA supplements may be considered in older patients with PKU unable to adhere to diet therapy. They have not been tested in children under the age of 11 years and are not advocated in pregnancy. This treatment is not used widely, there are limited LNAA supplements available and no preparation is on ACBS prescription in the UK. There is little advantage in using LNAA in patients who are adherent to their diet therapy.
Glycomacropeptide
Glycomacropeptide (GMP) is a low phenylalanine protein source used as an alternative to L-amino acids in the treatment of PKU. GMP constitutes 15%–20% of the protein found in whey. Pure GMP contains 47% (weight/weight) of indispensible amino acids. It contains no aromatic amino acids including phenylalanine, tyrosine, tryptophan, histidine, arginine and cysteine. It is also low in methionine and leucine but contains elevated amounts of threonine and isoleucine. To ensure GMP is low in phenylalanine it undergoes special processing through chromatography, ultrafiltration/diafiltration and lyophilisation to reduce phenylalanine content to <2 mg/g of GMP [133]. Commercially adapted GMP protein substitutes that are currently available are high in isoleucine and threonine (compared with phenylalanine free L-amino acids) and have required supplementation with some L-amino acids including tyrosine, tryptophan, histidine and leucine [134].
There is some evidence that GMP, when given to PKU mice, competitively inhibits phenylalanine transport into the brain and PKU mice show comparable growth and reduced concentrations of plasma and brain phenylalanine compared with a conventional amino acid source [135]. The concentrations of isoleucine and threonine in plasma showed a significant two- to three-fold increase in the GMP fed mice. Human research is very limited and there is very little long term outcome data, no information about nutritional status or any information outlining GMP’s safety in children <11 years of age. Short term data, however, in older patients suggest fasting phenylalanine concentrations were significantly lower with the GMP compared with an L-amino acid supplement. Blood urea nitrogen was also significantly lower with GMP [136]. There is some suggestion that it lowers postprandial concentrations of the appetite stimulating hormone ghrelin and may help promote satiety [137]. Low phenylalanine GMP products that can be used to replace phenylalanine free L-amino acid supplements are obtainable in the USA; they are not available in the UK on ACBS prescription.
Biopterin-responsive phenylalanine hydroxylase deficiency
Sapropterin dihydrochloride is the biologically active synthetic form of tetrahydrobiopterin (BH4), the naturally occurring essential cofactor for the enzyme PAH [138]. Some mutant PAH enzymes show enhanced activity in the presence of pharmacological doses of sapropterin [139], so it is effective in a subset of patients mainly with mild or moderate phenotypes of PKU [140]. Sapropterin has been demonstrated to lower blood phenylalanine concentrations [141] and improve phenylalanine tolerance [142, 143]. It is an oral medication given as an adjunctive therapy with a ‘relaxed’ low phenylalanine diet, or it may replace diet therapy depending on the individual response to the drug [144]. Approximately 20%–40% of patients achieve ≥30% reduction in blood phenylalanine concentration with a BH4 loading test, with a dose of 20 mg/kg [140, 145]. The dose of sapropterin is titrated to individual patient requirements (5–20 mg/kg) [146]. Long term data describing blood phenylalanine control, neuropsychological outcome and nutritional status are limited. There is evidence to suggest that even with sapropterin patients still require careful long term monitoring to prevent loss of dietary control associated with poor dietary adherence, excessive natural protein intake and catabolism associated with infections [147].
Phenylalanine ammonia lyase enzyme substitution therapy
Phenylalanine ammonia lyase (PAL; EC 4.3.1.5) is an enzyme that degrades phenylalanine into trans-cinnamic acid and ammonia [148]. It does not require a cofactor. Although its administration lowered blood and brain phenylalanine concentrations in mice [149], its repetitive and extended exposure induced an immune response [150]. This has led to the development of a pegylated phenylalanine ammonia lyase (PEG PAL) to aid the suppression of immune recognition with repeated PAL injections [151].
The use of PEG PAL is being investigated in humans. The drug Phenylase™ (PAL) (Biomarin Pharma) is given by subcutaneous injections to patients >16 years with PKU. In Phase 2 clinical trials it has been administered to 25 subjects for 1 year and it lowered blood phenylalanine on average by 68% compared with pretreatment levels, with all 25 patients maintaining blood phenylalanine concentrations <600 µmol/L. The main principal adverse reaction reported is hypersensitivity type reactions (injection site reactions, disseminated skin reactions or joint pain). This occurs during the initial dosing of the drug [152]. In theory tyrosine supplementation should still be required [148].
Genome targeted gene therapy
Liver directed gene therapy, by using recombinant adenoassociated virus (AAV) vectors, has corrected hyperphenylalaninaemia to normal in PAH deficient mouse models. AAV vectors produce minimal immune response and give longer lasting effects compared with the initial vectors used in gene therapy [153]. This does not, however, lead to permanent correction of liver PAH deficiency. Skeletal muscle has also been successfully used as an alternative target for gene therapy in PAH deficient mouse models [154].
Pharmacological chaperones
There is progress in identifying alternative pharmacological chaperones to BH4 that will enhance the thermal stability and activity of PAH [155, 156].
Maple syrup urine disease
Maple syrup urine disease (MSUD) is a neurotoxic, autosomal recessive disorder of branched chain amino acid (leucine, isoleucine and valine) catabolism. The first step in the catabolism of branched chain amino acids (BCAA) is their transamination to form the branched chain keto acids (BCKA): 2-oxoisocaproic acid (leucine), 2-oxo-3-methylvaleric acid (isoleucine) and 2-oxoisovaleric acid (valine) [157] (Fig. 17.9).
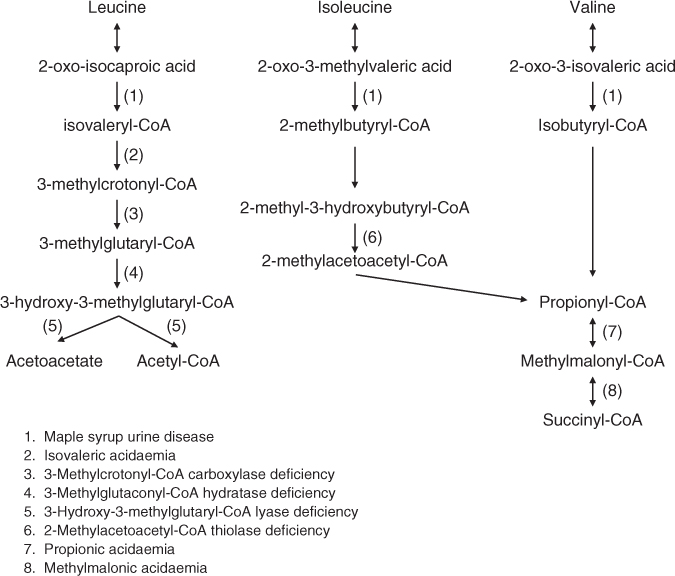
Figure 17.9 Selected inborn errors of the catabolic pathways of branched chain amino acids.
In MSUD, there is a deficiency of the multienzyme α-ketoacid dehydrogenase (BCKAD; EC.1.2.4.4.) complex, which is required for the second shared step in the degradation of BCKA. BCKAD is composed of three enzymes: a 2-oxo-decarboxylase (E1) that requires thiamin as a cofactor, dihydrolipoyl acyltransferase (E2) and dihydrolipoyl dehydrogenase (E3).
Deficiency of BCKAD causes accumulation of high concentrations of leucine, valine, isoleucine and alloisoleucine as well as their corresponding BCKA in plasma and cerebrospinal fluid. Neurological sequelae are present in most patients. Although increased plasma concentrations of leucine and/or its keto acid 2-oxoisocaproic acid are considered the main neurotoxins [158] the mechanisms underlying the brain damage are not well established. Brain injury in MSUD is also associated with demyelination [159, 160], neurotransmitter disturbances, induction of oxidative stress [161], apoptosis and energy deficit [162]. The neurotoxicity of leucine may be associated with its ability to inhibit transport of other LNAA across the blood–brain barrier (BBB), reducing the brain supply of amino acids such as tryptophan, tyrosine, phenylalanine, valine and threonine [163, 164]. There appears little apparent toxicity associated with increased plasma concentrations of isoleucine or valine [158].
Prevalence
MSUD has an estimated frequency of approximately 1 in 116 000 [165]. In the Mennonite populations of Pennsylvania, Kentucky, Indiana and Wisconsin, USA, it occurs in approximately 1 in 176 newborns [158]. The frequency is also high in the Ashkenzi Jewish population and in countries where consanguineous marriage is common [160].
Classification
MSUD is a genetically and phenotypically heterogeneous disorder of variable clinical severity from a severe classical form to a milder variant. Five clinical and biochemical phenotypes are known (Table 17.9).
Table 17.9 Clinical phenotypes of MSUD [163, 166–168]
| Type and BCKAD (% of normal activity) | Age of onset |




