Highlights
- •
Bacterial infection of the prostate may drive inflammation and cellular plasticity that contributes to the development of prostate cancer.
- •
About 1 mechanism whereby the gut microbiome is associated with prostate cancer risk is via the conversion of dietary precursors into carcinogenic metabolites.
- •
Prostate cancer treatment resistance could be mediated via direct drug metabolism by the commensal gut microbiota as well as androgen metabolism by gut microbiota.
Abstract
Complex relationships between the human microbiome and cancer are increasingly recognized for cancer sites that harbor commensal microbial communities such as the gut, genitourinary tract, and skin. For organ sites that likely do not contain commensal microbiota, there is still a substantial capacity for the human-associated microbiota to influence disease etiology across the cancer spectrum. We propose such a relationship for prostate cancer, the most commonly diagnosed cancer in males in the United States. This review explores the current evidence for a role for the urinary and gut microbiota in prostate cancer risk, via both direct interactions (prostate infections) and long-distance interactions such as via the metabolism of procarcinogenic or anticarcinogenic dietary metabolites. We further explore a newly recognized role of the gut microbiota in mediating cancer treatment response or resistance either via production of androgens and/or procarcinogenic metabolites or via direct metabolism of anticancer drugs that are used to treat advanced disease. Overall, we present the current state of knowledge relating to how the human microbiome mediates prostate cancer risk, progression, and therapy response, as well as suggest future research directions for the field.
Introduction
The human microbiome is comprised of a diverse and multifaceted community of microorganisms that inhabit all areas of the human body exposed to the outside world. This includes the entirety of the gastrointestinal (GI) tract, the skin, the upper respiratory tract, and parts of the genitourinary system. Whereas the human microbiota play a pivotal role in maintaining health and various physiological processes, a complex interplay is also known to exist between the human microbiome, microbial metabolites, dietary components, and the development or progression of cancer. Furthermore, an increasingly recognized function of the human microbiota is in modulating drug metabolism [ ] and cancer treatment response.
Many of the initial discoveries linking the microbiome and diet to cancer risk and progression arose in the field of GI cancers (namely colorectal and gastric). This is not surprising due to the abundant and complex microbial communities that exist in direct contact with human epithelium along the GI tract. Furthermore, the GI microbiome is responsible for the vast majority of the metabolism of dietary components for the human body [ ]. As such, the capacity of the GI tissues to be directly exposed to, for example, deleterious metabolites or dietary carcinogens is greatest in this anatomic location [ ]. Likewise, there is a known association between inflammation driven by the gastric pathogen Helicobacter pylori and the development of gastric cancer [ ], as well as suspected associations between colonic pathogens and the development of colorectal cancer [ ] for which mechanistic data continues to accumulate.
Relationships between the human-associated microbiota and cancer etiology likely exist for other areas of the human body, yet have been harder to study due to the potential for both direct and “long distance” indirect interactions between where the microbes reside and where the cancer arises [ ]. For example, unlike the GI tract, the human prostate likely does not contain commensal microbiota; however, the capacity of the human commensal microbiota in the urinary tract as well as the GI tract to influence all aspects of the prostate cancer spectrum, from cancer initiation through metastatic disease, is unquestionable ( Fig. 1 ). This review explores direct interactions of the urinary microbiota in prostate cancer risk as well as potential long-distance interactions between the GI microbiota and their associated metabolites with prostate cancer risk, progression, and treatment response. We likewise explore the role of the GI microbiota and dietary metabolites in promoting prostate carcinogenesis. Finally, we present evidence that the GI microbiota contribute to prostate cancer treatment resistance, either via production of androgens and other procarcinogenic metabolites or via direct metabolism of drugs that are used to treat advanced disease.
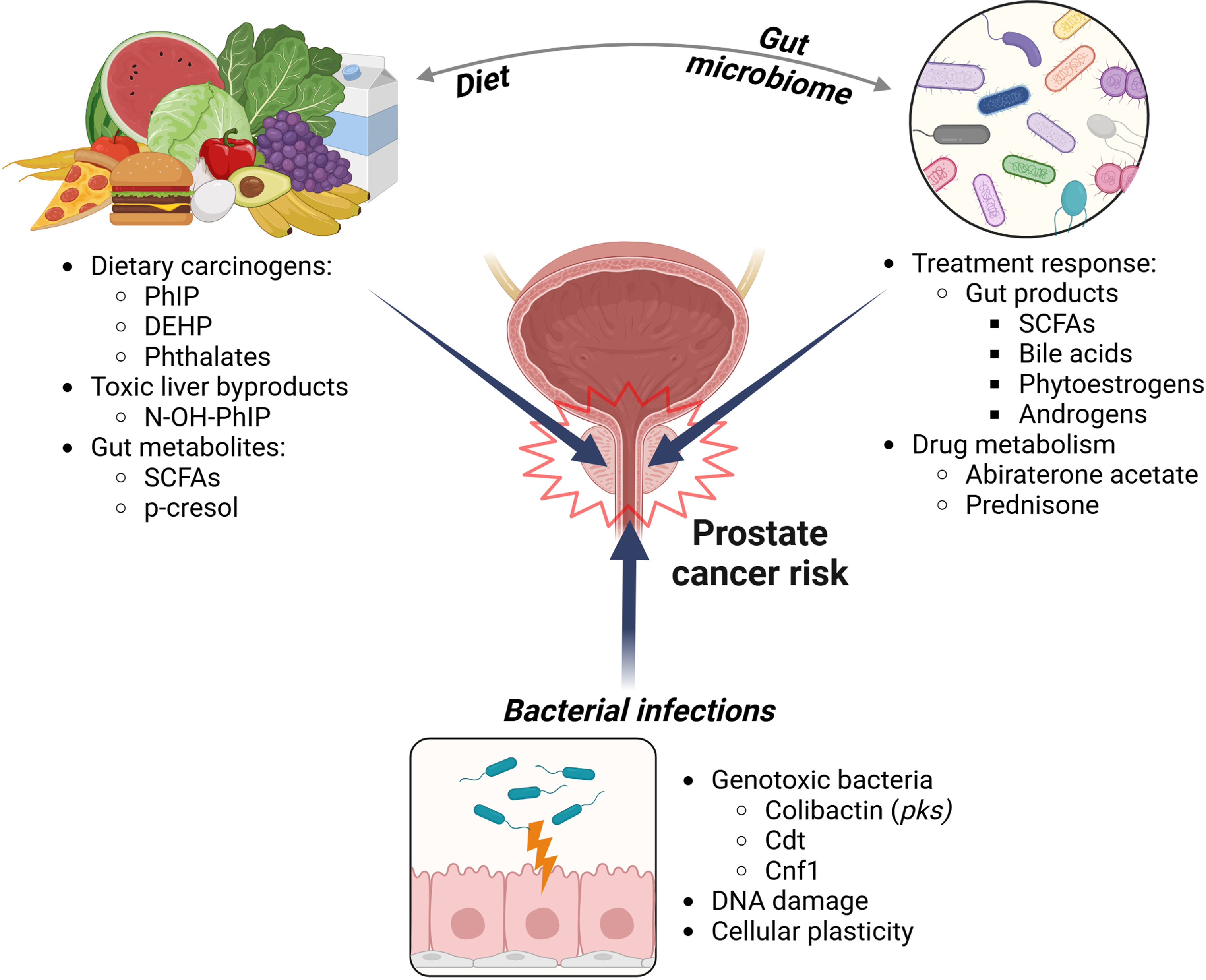
Infections and the urinary microbiome in prostate cancer risk
Within the past 10 years, a commensal urinary microbiome has been well characterized and its dysbiosis (a pathogenic imbalance of microbial communities) is potentially associated with multiple genitourinary cancers [ ]. Despite the evidence for a microbial presence within the urinary tract, researchers have found little evidence for a normal, commensal microbiome in the prostate [ ]. Nevertheless, the prostate can suffer from bacterial infections that contribute to acute and chronic bacterial prostatitis. Bacteria associated with acute prostatitis include Escherichia coli, Pseudomonas aeruginosa , and other known urinary pathogens, implicating a urinary source [ ]. In the context of prostate cancer, proinflammatory bacteria and pathogens associated with urinary tract infections (UTIs) were found to be more abundant in the urine of individuals undergoing biopsy for prostate cancer that were prostate cancer positive vs. those with a negative biopsy [ ]. However, it remains unclear whether UTI pathogens play a role in promoting or initiating prostate carcinogenesis, or if their presence occurs as a result of prostate cancer. Epidemiological studies deliver mixed results on whether UTIs confer a greater risk of prostate cancer later in life [ , ]. Individuals with a history of prostatitis (including all prostatitis, as well as acute and chronic bacterial prostatitis) have a slightly elevated risk for developing prostate cancer (odds ratio = 1.6:1.9) [ , ]. A major drawback of epidemiological-based studies is the reliance on retrospectively clinically diagnosed UTIs or prostatitis, potentially overlooking bacterial species associated with subclinical infection that may be associated with long-term risk of prostate cancer. Despite the mixed evidence for prior UTI or prostatitis and prostate cancer risk, there are several potential mechanisms whereby infection and inflammation due to uropathogenic bacteria may contribute to carcinogenesis in the prostate.
Mechanisms for direct DNA damage to prostate epithelial cells by bacterial genotoxins. Genotoxins are chemicals, peptides, or biomolecules that cause damage to DNA or chromosomes. Bacterial genotoxins are a type of virulence factor used to establish infection and outcompete other bacterial species. In a human host, bacterial genotoxins can initiate carcinogenic mutations through the generation of single-stranded (SSB) and double-stranded (DSB) DNA breaks [ ], and also via forming DNA adducts that can induce mutations [ ]. Three key genotoxins are implicated in prostate cancer etiology: colibactin, cytolethal distending toxin, and cytotoxic necrotizing factor.
Colibactin. The colibactin genotoxin is a polyketide-nonribosomal peptide hybrid that is encoded by bacteria carrying the pks genomic island [ ]. Colibactin directly damages DNA by creating intrastrand crosslinks and inciting torsional stress, resulting in DSB [ ]. Colibactin is a well-studied genotoxin that creates a unique DNA damage motif that is enriched in colorectal cancers and, interestingly, is also found in cancers of the urinary tract [ , ]. Genomic damage induced by colibactin has likewise been implicated in prostate cancer [ ]. We have previously demonstrated that colibactin-producing E. coli initiate DSB in prostate cell lines and lead to chromosomal rearrangements, indicating that colibactin may play a causative role in initiating common oncogenic gene fusions found in prostate cancer [ ] ( Fig. 2 ).
Cytolethal distending toxin (cdt). Cdt is a bacterial virulence factor found in pathogenic strains of Gram-negative bacteria. The catalytic subunit CdtB displays DNase-like activity and generates SSBs that are ultimately converted into DSBs during S-phase [ ]. Cdt is produced by several Gram-negative bacterial species ( E. coli, Campylobacter sp. , Helicobacter sp., etc.), yet retains similar biological activity among species [ ]. To date, there have been few direct studies investigating cdt genotoxicity on normal prostate epithelial cells. Interestingly, mice with dysregulated WNT signaling (Apc Min/+) are more susceptible to prostate cancer after gastrointestinal infection with cdt+ Helicobacter hepaticus [ ].
Cytotoxic necrotizing factor (cnf1). Cnf1 is a prevalent virulence factor of uropathogenic E. coli . In contrast to colibactin and cdtB, cnf1 does not induce direct DNA damage and instead modifies a GTP-binding protein known as Rho, resulting in constitutive activation of pathways involved in migration, cell division, and polarity [ ]. In relation to prostate cancer, cnf1 was found to promote the migration and invasiveness of prostate cancer cell lines in vitro [ ], however few studies have investigated a potential role for cnf1 in carcinogenic transformation.
Prevalence of genotoxic bacteria in association with prostate cancer. Genotoxic bacteria have been identified in urine from individuals with acute bacterial prostatitis [ ] as well as radical prostatectomy specimens harboring bacterial infections [ ]. In a prior study, the prevalence of genotoxin-carrying bacteria in urine samples taken prior to prostate biopsy was reported as 8% (25/311) [ ]. Of the 25 samples that were positive for bacterial genotoxin genes, the colibactin genotoxin was the most frequently identified ( clbN, clbB ; 19/25), followed by cytolethal distending toxin ( cdtB ; 10/25), and cytotoxic necrotizing factor toxin ( cnf1 ; 7/25) [ ]. The authors of this study argue that the prevalence of urinary genotoxins is low and does not correspond to a prostate cancer diagnosis [ ]. However, the study carries some important limitations. First, more than 1 time point is needed to appropriately survey a casual role of genotoxic bacteria in prostate cancer diagnosis. Likewise, the study does not capture prior exposure that could have contributed to prostate cancer initiation (possibly many years prior) since urine samples were only examined at the time of prostate cancer diagnosis. Selection bias is another potential concern, since all of these individuals were undergoing evaluation for suspicion of prostate cancer, and likely do not represent the urinary genotoxin prevalence in the widespread population. However, the prevalence of bacteria carrying these 3 genotoxins in the urinary tract and as potential actors in bacterial prostatitis is supported by another study that also identified colibactin ( clbA-N ), cdtB, and cnf1 in urinary E. coli isolates from individuals suffering from acute bacterial prostatitis [ ]. The colibactin genotoxin was also the most frequently identified in this study (13/18 isolates) [ ].
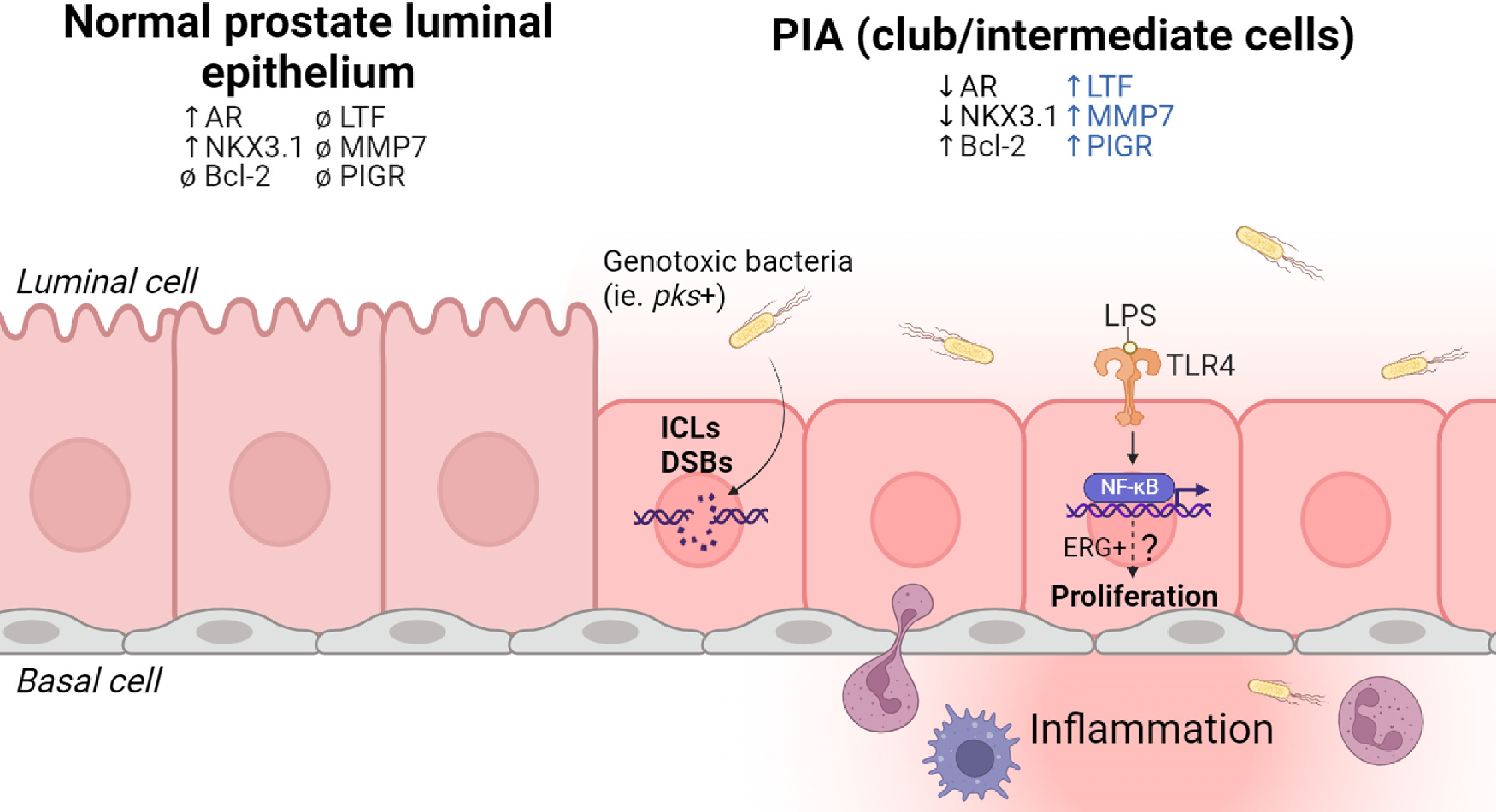
Bacterial-mediated activation of TLR4/NF-kB signaling. The innate immune response to bacteria at mucosal surfaces is mediated in part by toll-like receptors (TLRs) that recognize pathogen-associated molecular patterns (PAMPs) in the environment. Prostate epithelial cells express TLRs and respond to bacterial by-products (e.g., lipopolysaccharide, LPS) through downstream activation of NF-kB and secretion of proinflammatory cytokines [ , ]. In prostate cancer, constitutive NF-kB activation may promote a more aggressive phenotype and is associated with metastasis and castration resistance in animal models [ , ]. However, it is unknown if persistent NF-kB activation similarly promotes malignant transformation of normal prostate epithelial cells. Chronic exposure to LPS and cognate TLR4/NF-kB activation may serve as a potential mechanism linking inflammation to carcinogenesis. LPS increases proliferation of the immortalized prostate epithelial RWPE cell line and renders the cells more resistant to apoptosis, an effect that is abrogated when TLR4 is silenced [ ]. We speculate that it is unlikely that the LPS-induced proliferation drives carcinogenesis in normal epithelial cells, but it could serve as a mechanism promoting the outgrowth of premalignant epithelial cells ( Fig. 2 ).
In support of this concept, LPS stimulation of TLR4 and NF-kB may provide a selective growth advantage to normal prostate epithelial cells containing TMPRSS2-ERG (T:E) gene fusions [ ]. When the immortalized PNT1a normal prostate epithelial cell line was transfected with common T:E gene fusion isoforms, NF-kB activation occurred that was TLR4-dependent [ ]. The relationship between TLR4 and the T:E gene fusion appears to be a feed-forward mechanism: activation of TLR4 promotes the transcriptional activation of ERG, and ERG promotes the expression of TLR4 [ ]. We speculate that bacterial-mediated activation of TLR4 may provide a selective advantage for rare ERG+ prostate epithelial cells in proliferative inflammatory atrophy (PIA) lesions [ ] by activating ERG-mediated migration, survival, and growth pathways. Nevertheless, there is a lack of direct evidence for whether chronic TLR4 stimulation promotes carcinogenic transformation of premalignant ERG+ prostate epithelial cells [ , ] compared to normal prostate epithelial cells.
Bacterial infection-driven inflammation and cellular plasticity. Cellular plasticity is the ability of cells to alter their molecular and histologic phenotype in response to environmental stressors. In the context of carcinogenesis, the development of cellular plasticity may represent advantageous alterations on the path to becoming a cancer cell [ ]. Cellular plasticity within normal prostate epithelial cells is not well characterized, but it may occur in response to an inflammatory microenvironment. A key example is cellular plasticity in prostate luminal epithelial cells within an inflammation-associated lesion known as PIA [ , ]. Within PIA lesions, luminal prostate epithelial cells display an altered morphology from typical tall columnar cells to shortened cuboidal epithelium [ ], and display a gene signature canonically associated with club cells of the bronchiolar epithelium [ ] as well as the prostatic urethra and early budding prostatic ducts [ ]. Expression of club cell genes, coupled with a distinct histological appearance, fits the definition of a plastic phenotype — the long-term consequences of which are unknown. Intriguingly, top genes expressed in the club gene signature (lactoferrin, matrix metallopeptidase 7, polymeric immunoglobulin receptor) all have roles in microbial defense and immunoregulation. Whether the cellular plasticity of prostate epithelial cells occurs in response to an initial bacterial infection, or whether it is a normal, regenerative response to disrupted homeostasis is yet to be determined. Likewise, the role of cellular plasticity of prostate epithelial cells in the carcinogenic process merits further active investigation.
Diet and the gut microbiome in prostate cancer risk
The influence of the microbiome on prostate cancer likely goes well beyond direct interaction with the urinary microbiota. For example, the importance of diet and the gut microbiome on human disease cannot be understated; both factors have been independently, as well as synergistically, linked to a variety of debilitating human diseases such as gastritis, inflammatory bowel disease, obesity, heart disease, and cancer. Research into the role of diet and the gut microbiome in cancer is still in its infancy, however compounding evidence has shown that high consumption of red meat, as well as harboring gut microbes that can metabolize ingested components and produce their own metabolites can be potent carcinogens that drive or worsen cancer. Additionally, factors previously unexplored such as leaky chemicals from the food manufacturing processes and plasticizers have not been fully investigated in cancer, but their abundance in food and gut contents pose a potent threat to human health and disease.
Conversion of dietary precursors into toxic/carcinogenic metabolites by the gut microbiome and prostate cancer promotion. The biological fate of a meal begins with it traveling down the esophagus into the stomach, where bile and other gastric acids break up the food particles and siphon them down into the intestines. Here, stomach components are further chemically and mechanically digested until they become waste and are excreted. The resulting soluble molecules that remain circulate through the blood, and often end up at the liver – the master filter that can chemically convert biomolecules into products that are excreted through urine. However not all dietary molecules are safe when digested, and not all liver conversion detoxifies food byproducts ( Fig. 3 ). A clear example that is relevant to prostate cancer is 2-Amino-1-methyl-6-phenylimidazo(4,5-b)pyridine (PhIP) – the most abundant heterocyclic amine found in high-temperature grilled meat, particularly red meat. PhIP is converted to the bioactive compound N-hydroxy-PhIP (N-OH-PhIP) by the liver enzyme CYP1A2 [ ]. N-OH-PhIP is subsequently acetylated or sulfonated to electrophilic compounds that form PhIP-DNA adducts [ , ]. Many epidemiological studies have shown through dietary surveys that higher grilled meat consumption (used as a proxy for PhIP intake) is associated with increased risk of prostate cancer [ , ]. Additionally, higher grilled red meat consumption in African-Americans, an elevated-risk group for prostate cancer, may be associated with increased risk of developing prostate cancer or high risk prostate cancer [ , ].
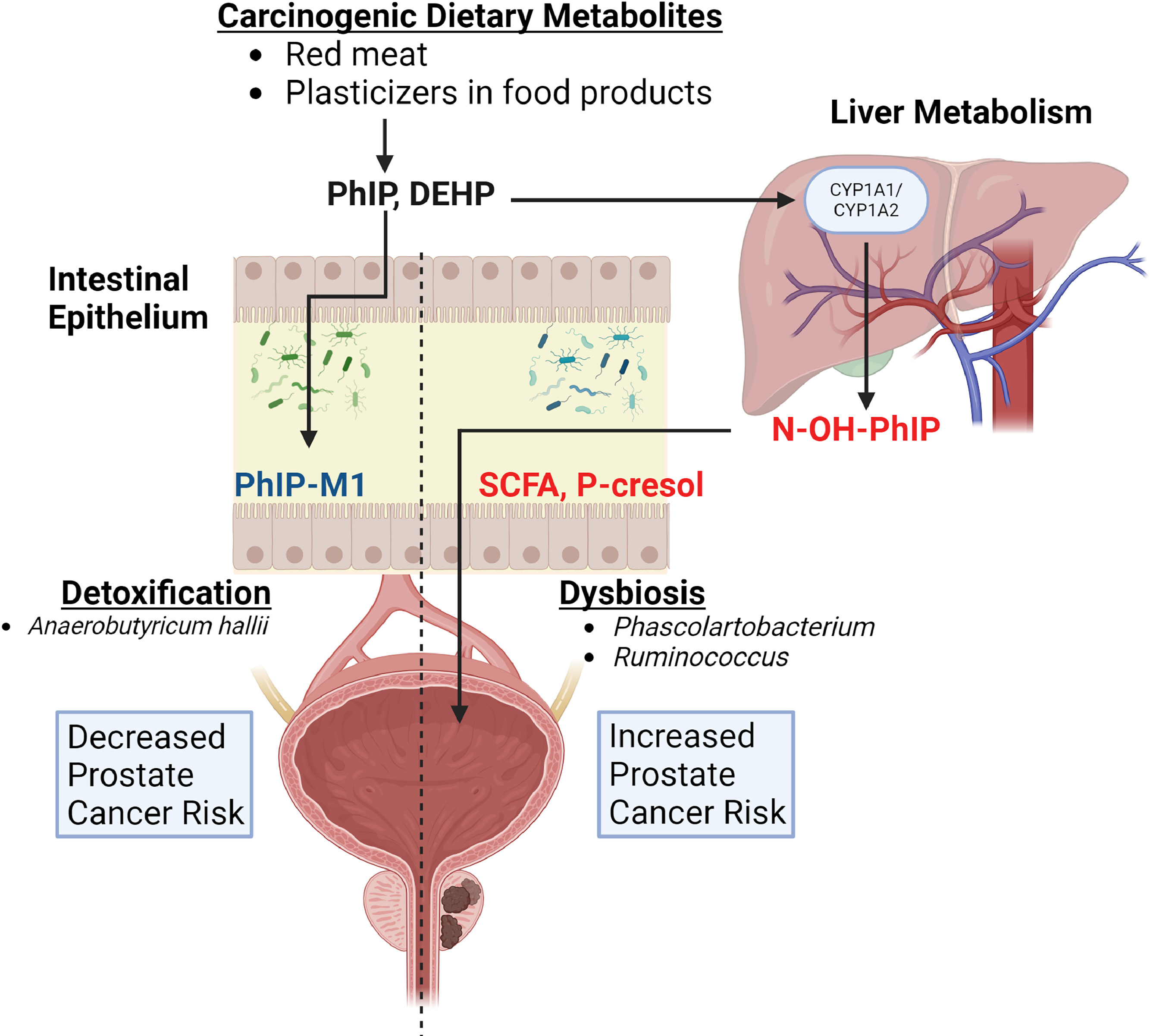
When PhIP is orally delivered to rats, preinvasive cancer lesions develop that are specific to the ventral lobe of the rat prostate [ , ]. In this setting, PhIP acts as both a cancer initiator as well as a tumor promoter, a phenomenon that may be mediated in part by PhIP-induced recruitment of mast cells and macrophages [ ]. Surprisingly, the co-occurrence of bacterial prostatitis with uropathogenic E. coli and PhIP consumption promoted an increase in precancerous lesions in the prostate, as well as an increase in PhIP-related invasive cancers at multiple other sites [ ]. This study also implicated a role for inflammation, both local and systemic, in PhIP-promoted tumorigenesis [ ]. Interestingly, studies have also found that the gut microbiome may play an important role in PhIP metabolism. The commensal gut bacterium, Anaerobutyricum hallii , can detoxify PhIP and convert it into a nontoxic byproduct called PhIP-M1 through its acrolein metabolic pathway [ ]. Fecal batch fermentation experiments demonstrated that supplementing with A. halli led to a significant reduction in PhIP levels. These findings, although in their infancy, have implications for preventative treatment strategies in reducing the risk of developing prostate cancer through dietary exposure.
Aside from PhIP, studies have demonstrated that other components of a “Western diet” (a diet that is high in calories, red meat, saturated fatty acids, and processed foods) increase the risk of developing lethal prostate cancer [ , ], and the gut microbiota likely play a fundamental role in mediating this relationship. For example, gut bacterial metabolism, along with host metabolism in the liver, is responsible for circulating levels of trimethylamine N-oxide (TMAO), a metabolite that is linked to cancer risk as well as cardiovascular disease [ ]. Likewise, a prior study found circulating TMAO levels to be positively associated with risk of aggressive prostate cancer [ ]. Similarly, the amino acid metabolites hippuric acid, p-cresol sulfate, and phenylacetylglutamine are produced by a combination of gut bacterial and host metabolism and are linked to cardiovascular disease [ ]. Serum samples from the randomized controlled Prostate, Lung, Colorectal, and Ovarian Cancer (PLCO) Screening trial [ ] were used to quantify levels of dietary TMAO precursors (choline, carnitine, betaine, gamma-butyrobetaine, crotonbetaine) as well as hippuric acid, p-crestol sulfate, and phenylacetylglutamine in individuals who did not develop prostate cancer or who developed nonlethal prostate cancer ( n = 519) vs. those who were diagnosed with prostate cancer and died of the disease ( n = 173). The authors found that individuals with elevated levels of choline, betaine, and phenylacetylglutamine (the product of gut microbial metabolism of the amino acid phenylalanine) were 2 to 3 times more likely to be diagnosed with lethal prostate cancer [ ]. The authors speculate that the relationship between phenylacetylglutamine and lethal prostate cancer may be mediated in part by its role in adrenergic signaling, which has also been shown to promote prostate cancer aggressiveness [ , ].
In addition to this endogenous conversion of dietary components to carcinogenic metabolites, the microbiome itself has also been shown to be altered by the consumption of a high-fat diet [ ]. Administration of a diet containing a high proportion of lard to the prostate-specific Pten knockout mouse model altered the gut microbiome in a manner that increased the number of short-chain fatty acid (SCFA)-producing bacteria [ ]. Furthermore, the resultant SCFA production contributed to worse prostate cancer outcomes in this animal model via reduction of insulin-like growth factor-1 (IGF1) levels systemically and within the prostate [ ]. SCFAs, including acetate, butyrate, and propionate, are the products of microbial anaerobic fermentation of indigestible polysaccharides, and are implicated in both procarcinogenic as well as protective anticancer roles [ ]. Yet, there are limited human studies of SCFAs in relation to prostate cancer. A study compared the fecal microbiota profile of 21 individuals sampled when they had hormone sensitive prostate cancer (HSPC) vs. when they developed castration resistant prostate cancer (CRPC) and reported an increased abundance of genera containing SCFA-producing bacteria such as Phascolarctobacterium and Ruminococcus in the CRPC samples [ ]. Furthermore, fecal microbiota transplant (FMT) from CRPC patients to the TRAMP mouse model increased gut acetate and butyrate levels and accelerated prostate cancer progression, possibly via a role in immunomodulation [ ]. It is therefore critical to understand the metabolomic pathways of both the liver as well as the gut microbes that break down food in order to develop novel targets against toxic and procarcinogenic metabolic byproducts.
Environmental exposure to estrogen-mimicking compounds such as phthalates through diet can result in dysbiosis and increased risk for prostate cancer. Dietary breakdown products are only a subset of potential carcinogens that can increase cancer risk. Chemicals from food manufacturing and packaging can also be ingested and alter the risk of developing cancer. Many epidemiological studies have shown that phthalates, an overarching term for a diverse group of chemicals that are used ubiquitously to stabilize and malleabilize many products, are associated with increased risk of cancer and other diseases [ ]. Furthermore, these chemicals are easily ingested and found in high-fat dairy products, fast foods, and packaged goods [ ], which are already known risk factors for certain cancers. As phthalates encompass a large family of chemicals, not all of them have been studied in detail to establish a positive relationship with cancer risk. However, sub studies on certain phthalates such as di(2-ethylhexyl) phthalate (DEHP), a high molecular weight (HMW) phthalate, and other HMW phthalates showed a correlation with increased prostate cancer risk [ ]. Another study found an association between DEHP, butyl-benzyl, and di-isobutyl phthalates and prostate cancer risk, specifically in obese individuals [ ]. Of interest, DEHP was previously shown to alter gut microbiota community structure and metabolism in mice, which resulted in the increased production of the potentially toxic metabolite p-cresol [ ]. Interestingly, the production of p-cresol was antagonistic to SCFA (butyrate) synthesis [ ], but the implications of this on increasing cancer risk, including prostate cancer, have not been studied. Overall, the studies to date strongly implicate the modification of dietary components by the gut microbiota in mediating prostate cancer risk, and this remains an area of active study.
The influence of the gut microbiome on prostate cancer growth and treatment response
Over the last several decades, new therapeutics have been developed to treat advanced prostate cancer, many of which target the androgen receptor (AR) signaling axis. While offering effective treatment options to some, most patients invariably develop resistance to treatment through unknown mechanisms. Some of the challenges to developing effective prostate cancer treatments may be in part due to variability within patients, disease characterization, and treatment protocols [ ]. We speculate that the interindividual variability in treatment response between patients could also be ascribed to differences in microbial communities that may both promote prostate cancer growth and interfere with treatment efficacy [ ]. Several studies highlight differences in the gut microbial composition from individuals with vs. without prostate cancer [ ] and those undergoing AR-targeted treatments [ , , , ]. Dysbiosis can directly and indirectly affect the distribution and richness of microbes and their products in the gastrointestinal and genitourinary tract, respectively. Shifts in gut microbial communities affects the production of SCFAs and bile acids, and even affects microbial metabolism responsible for enteric androgen synthesis.
Antibiotic use and prostate cancer treatment response. Multiple studies suggest that broad spectrum antibiotic use (which eliminates core members of the gut microbiota) is associated with decreased efficacy of systemic therapy for advanced cancer. The most mature data for this is in the demonstration of decreased efficacy of immune checkpoint inhibitors (ICI) in immunotherapy-responsive cancers such as melanoma, non-small cell lung cancer, and renal cell carcinoma [ ]. This particular association between antibiotic use and decreased efficacy of immunotherapy has not been studied in advanced prostate cancer, and prostate cancer is typically not responsive to ICI. A study that included 3,011 prostate cancer patients demonstrated that antibiotic use prior to non-ICI systemic therapy was not associated with poorer cancer survival, whereas as a strong association was found in breast and lung cancer [ ]. Another study reported that individuals with chemotherapy-naïve metastatic CRPC who received ≥ 30 days of antibiotic treatment during systemic therapy with oral drugs such as abiraterone acetate had prolonged overall survival and radiographic progression-free survival [ ]. Whereas the mechanism behind antibiotic usage and response to systemic therapy in prostate cancer remains unknown, it could potentially relate to androgen production by gut flora, as discussed below.
SCFAs in prostate cancer promotion and treatment response. In the early 1990s, SCFAs were considered differentiating agents that were able to diminish the ability of prostate cancer cells to form tumors in mice [ , ]. Since then, several mechanisms of action have been postulated for these metabolites. SCFA levels can be attributed to changes in diet, host diseases and their treatments, as well as the levels of microbes responsible for their production. Studies have shown that certain SCFA-producing bacteria and their metabolites are increased in individuals with high grade prostate cancer [ ]. This increase in SCFA-producing gut bacteria leads to a surge of SCFAs in the gut which, as previously mentioned, has been shown to promote prostate cancer growth via the IGF-1 signaling pathway [ ]. Additionally, studies demonstrate that SCFAs affect cell epigenetics by affecting histone deacetylase (HDAC) activity [ ], which is known to modulate cellular events such as cell migration, proliferation, and survival.
Due to the potential for SCFAs to drive prostate cancer growth, it is plausible that SCFAs may also influence prostate cancer treatment response. Indeed, SCFAs are reported to influence cancer therapy. In a study focusing on patients with solid tumors, longer progression-free survival upon treatment with pembrolizumab was associated with higher fecal SCFAs (acetic acid, propionic acid, butyric acid, valeric acid) and plasma isovaleric acid [ ]. Somewhat paradoxically, higher levels of circulating butyrate and propionate were also associated with resistance to CTLA-4 blockade, which may be mediated by higher proportions of regulatory T (Treg) cells [ ]. Continuous exposure to butyrate in mice restrained anti-CTLA-4-induced up-regulation of CD80/CD86 on dendritic cells and ICOS on T cells and the accumulation of tumor-specific T cells and memory T cells [ ]. Pelvic radiation therapy for prostate cancer treatment also affects the gut bacterial population of SCFAs producers, which was previously shown to mediate increased gastrointestinal side effects [ ]. However, there is contradicting evidence regarding SCFAs and radiotherapy in cancer progression, thus additional mechanistic studies are needed. These studies will need to address topics such as the effect of cancer treatments on SCFAs producers and their metabolites (circulating vs. fecal) before and after treatment, as well as to define the molecular mechanism of action considering the variations in the gut microbiota and their metabolites as a community.
Gut microbial bile acids inhibit prostate cancer proliferation. Primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA), are steroid acids that are produced in the liver and secreted into bile. In the gut, primary bile acids are converted to secondary bile acids, deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA), by the gut commensal microbiota. The suspected role of bile acids in cancer etiology has evolved throughout the years. Initially, bile acids were thought to be tumor promoters [ ], but most recently they have also been considered as tumor suppressor molecules depending on their concentration and type [ ]. Based on the bile acid hydrophobicity (LCA> DCA> CDCA> UDCA), they can act differently depending on the environmental stimulus. In relation to prostate cancer, LCA inhibits proliferation of the androgen dependent LNCaP prostate cancer cell line, while inducing cell death in both LNCaP and androgen independent PC3 cells [ ]. UDCA inhibits the growth of the androgen independent DU-145 prostate cancer cell line via the Tumor Necrosis Factor Related Apoptosis-Inducing Ligand (TRAIL) signaling pathway [ ]. CDCA and DCA can promote antitumor phenotypes by destabilizing HIF-1α subunits, suppressing cancer progression-associated phenotypes such as clonogenic growth, invasion, and migration in DU-145 cells [ ].
Interestingly, bile acids modulate the gut microbiota, and they are linked to microbial metabolism. Whereas there are limited studies that explore the role of bile acids in prostate cancer treatment response, androgen deprivation therapy (ADT) with a gonadotropin releasing hormone (GnRH) agonist was found to increase the circulating levels of bile acids relative to levels measured prior to treatment [ ]. Whether this increase in circulating bile acids with ADT can be attributed to ADT-induced shifts in gut microbial populations is yet to be determined. Likewise, the influence of the increase in bile acids on ADT treatment response or side effects, such as weight gain, lowered insulin sensitivity, and dyslipidemia is currently unknown.
Microbial metabolism of phytoestrogens suppresses prostate cancer growth. The gut microbiota play an important role in the metabolism of genistein and daidzein, soy isoflavones derived from dietary intake of soy. Genistein, daidzein, and some of their secondary metabolites are found to be protective against cancer [ ]. However, interindividual variations play an important role in the microbial metabolism of phytoestrogens [ ]. For example, only a third of the human population carries the gut bacterial species required for the conversion of daidzein to equol, a metabolite that has been found to inhibit prostate cancer growth in vitro and in vivo [ ]. Equol was shown to suppress cell proliferation and induce cell death in LNCaP cells, potentially by suppressing AR [ ] and by activating FOXO3a via Akt-specific pathways [ ]. Equol selectively binds to estrogen receptor (ER) beta, and is characterized as both a selective ER modulator and an ER agonist [ ]. Although equol does not directly bind to the androgen receptor (AR), it may bind to dihydrotestosterone (DHT) and prevent the stimulatory effect of DHT on prostate cell growth [ ]. Likewise, equol may induce direct degradation of AR via an S-phase kinase-associated protein 2 (Skp2)-mediated mechanism [ ]. Although not well studied, this relationship between equol and AR may hold important implications regarding prostate cancer treatment response, especially to AR-targeted therapies.
Androgen synthesis by gut microbiota and prostate cancer treatment response. The gut microbiota have been shown to influence circulating levels of testosterone (T) and dihydrotestosterone (DHT) in rodents [ , ]. The gut microbiota are involved in deglucoronidation of T and DHT predominantly in the large intestine, whereas free DHT is mostly found in the distal intestine at higher levels than in the circulation [ ]. However, the capacity for microbial deglucoronidation can be affected by interindividual variation of the gut microbiota. Furthermore, direct androgen metabolism by the gut microbiota has been ascribed to several known bacterial enzymes that are capable of metabolizing precursors into potent androgens ( Table 1 ) [ ]. Animal studies demonstrate that germ-free mice that lack intestinal microbiota have very low distal intestine free DHT levels [ ]. Likewise, animal studies demonstrate that antibiotic treatment may even delay the emergence of resistance to ADT therapy [ ]. The specific bacterial enzymatic pathways that are involved in gut androgen production that may contribute to endocrine resistance are currently unknown.
Related posts:
 Tumor involvement of the trigone and urethra at the time of robot-assisted radical cystectomy is associated with adverse oncological outcomes
Tumor involvement of the trigone and urethra at the time of robot-assisted radical cystectomy is associated with adverse oncological outcomes
 5α-reductase inhibitors with or without alpha-blockers and risk of incident upper tract urothelial carcinoma in men with benign prostatic hyperplasia: Analysis of US insurance claims data
5α-reductase inhibitors with or without alpha-blockers and risk of incident upper tract urothelial carcinoma in men with benign prostatic hyperplasia: Analysis of US insurance claims data
 Quantification of micropapillary component on transurethral resection is associated with likelihood of occult lymph node metastasis at radical cystectomy
Quantification of micropapillary component on transurethral resection is associated with likelihood of occult lymph node metastasis at radical cystectomy
 Causal relationship between bladder cancer and gut microbiota contributes to the gut-bladder axis: A two-sample Mendelian randomization study
Causal relationship between bladder cancer and gut microbiota contributes to the gut-bladder axis: A two-sample Mendelian randomization study
 Genetic variations related to the prostate cancer risk: A field synopsis and revaluation by Bayesian approaches of genome-wide association studies
Genetic variations related to the prostate cancer risk: A field synopsis and revaluation by Bayesian approaches of genome-wide association studies
 Clinical significance of PSA dynamics in castration-sensitive prostate cancer treated with ARSI doublet therapy: A multicenter study
Clinical significance of PSA dynamics in castration-sensitive prostate cancer treated with ARSI doublet therapy: A multicenter study
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


