Diagnostic Immunology
Thomas A. Fleisher
Joao Bosco Oliveira
Jack J. H. Bleesing
INTRODUCTION
Techniques for evaluation of immunologic function have expanded dramatically in recent years as understanding of the immune system increases. These studies are useful for the evaluation of patients with suspected immune deficiency disorders and autoimmune, allergic, or malignant diseases. The chapter is directed at general concepts and appropriate applications of methods used to characterize and quantify immunoglobulins and specific antibodies; evaluate lymphocyte phenotype and function; study neutrophil function, assay complement, as well as molecular methods used to enhance these other studies and make a definitive genetic diagnosis.
I. QUALITATIVE AND QUANTITATIVE MEASUREMENT OF IMMUNOGLOBULINS AND SPECIFIC ANTIBODIES
Immunoglobulins are glycoproteins secreted by differentiated B lymphocytes (plasma cells). Specific immunoglobulins (antibodies) are normally produced following antigenic stimulation, yielding a variety of antibody molecules, all of which react with varying strengths to the specific antigen. This type of response is referred to as polyclonal because it involves multiple B-cell clones. The basic molecular structure of the immunoglobulin consists of two identical heavy chains (α, δ, ε, γ, or µ) and two identical light chains (k or l) that together form a monomer (see Chapter 1). There are five different classes or isotypes of immunoglobulin that are determined by the heavy chain used in the molecule (e.g., immunoglobulin G [IgG] contains two identical g heavy chains paired with two identical k or l chains). The Fab portion of the molecule provides the antigencombining sites, and the Fc portion contains the other sites of biologic function, such as complement component binding. IgG, the most prevalent immunoglobulin in serum, is found as a monomer and constitutes the major antibody class produced in secondary antibody responses. Both IgE, the antibody of the allergic response, and IgD, a principal B-cell surface immunoglobulin, are also present in serum as monomers but are found in very low concentrations. IgA is found in serum and secretions as a dimer, together with a J chain and secretory component. The secretory component is synthesized by mucosal epithelial cells and joined to the dimeric IgA molecule as it passes through these cells. IgM, normally present in serum as a pentamer, is the immunoglobulin class produced early during a primary antibody response to an antigen.
Evaluation of immunoglobulins consists of qualitative and quantitative tests that are useful for evaluating immunoglobulin classes and their subclasses and/or antigen-specific antibody. Age-dependent change in the levels of immunoglobulins must be taken into account when interpreting results.
Decreases in the level of a serum protein, such as an immunoglobulin, can result from impaired synthesis, altered utilization, and/or increased loss. Immunoglobulin molecules of a particular class or subclass all share the same chemical structure, are recognized by a common structure in the constant region of the heavy (or light) chain, and, as such, can behave similarly in an immunoassay. The variable region of a specific antibody reacts with an antigenic epitope that is part of intact molecular structure of the antigen, conferring the “specific” nature of the antibody. Binding of the antigen to specific antibodies is also defined by a qualitative aspect of strength of the binding between antibody and antigen (defined by affinity for one interaction and avidity for the combined affinities in the case of multiple epitopes), making quantitative measurement of specific antibodies more difficult.
A. Qualitative Assessment (Electrophoretic Methodologies)
1. Zone electrophoresis. This technique allows for the separation of proteins based on electrical charge. The sample (solution) to be tested is placed on a buffer-saturated support medium (e.g., paper, agarose) and subjected to an electropotential gradient. Serum electrophoresis normally yields five bands consisting of albumin, alpha-1, alpha-2, beta, and gamma globulin fractions (Fig. 20-1). These bands can be assessed with a densitometer that generates a tracing from which the relative percentage of each fraction is determined. Immunoglobulins normally fall in the gamma globulin band, although they also migrate into the beta and alpha-2 globulin bands. Zone electrophoresis can be performed on other body fluids, including cerebrospinal fluid (CSF) and urine. This semiquantitative technique is useful for assessing total protein status and can be used to screen for monoclonal immunoglobulins, although this technique may miss low-level monoclonal antibodies seen in early myelomas.
2. Immunoelectrophoresis (IEP) is a two-step method in which proteins are electrophoretically separated in a gel and then antiserum, directed against one immunoglobulin heavy chain (α, δ, ε, γ, or µ) or one light chain (k or l), is loaded into a trough in the gel, and is allowed to diffuse
into the gel containing these separated proteins. The antibody combines with the appropriate protein (immunoglobulin), resulting in the formation of a precipitin arc(s) of the antibody-antigen complex in the area of the gel where antigen and antibody concentrations are at equivalence. This has been used primarily for the characterization of monoclonal immunoglobulins, but in the clinical laboratory, this method is now largely replaced by immunofixation electrophoresis.
into the gel containing these separated proteins. The antibody combines with the appropriate protein (immunoglobulin), resulting in the formation of a precipitin arc(s) of the antibody-antigen complex in the area of the gel where antigen and antibody concentrations are at equivalence. This has been used primarily for the characterization of monoclonal immunoglobulins, but in the clinical laboratory, this method is now largely replaced by immunofixation electrophoresis.
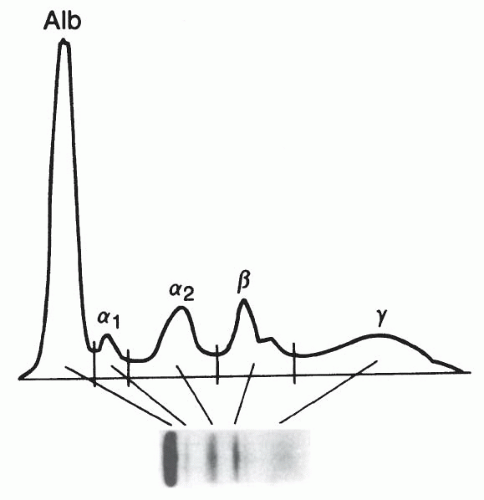 Figure 20-1. Zone electrophoresis. The electrophoretic pattern and densitometric tracing demonstrate the five major bands (albumin plus alpha-1, alpha-2, beta, and gamma globulins) from a normal serum sample. |
3. Immunofixation electrophoresis (IFE) uses zone electrophoresis, followed by overlaying of the electrophoretically separated proteins with antibodies directed against specific immunoglobulin heavy or light chains. This results in immunoprecipitation of the antibody-antigen complex (at equivalence), which can then be stained (e.g., imido black) and visualized (Fig. 20-2). Polyclonal immunoglobulins give a diffuse band, a monoclonal immunoglobulin produces an intense narrow band typically within the diffuse band in the background, and oligoclonal immunoglobulins yield multiple bands of increased intensity against the background. This technique is easier to interpret and more sensitive than IEP, though it is not quantitative. The test currently represents the standard clinical laboratory approach for identification of monoclonal or oligoclonal immunoglobulins.
B. Quantitative Assessment (Precipitin Methodology)
1. Double gel diffusion is another semiquantitative test that evaluates the relationship between specific antibodies or antigens in solutions. Comparison between a reference material and unknown serum allows a comparative assessment for identity, partial identity, or nonidentity. Although it lacks the detection sensitivity of many quantitative methods, the test is technically easy, can be performed with antigen preparations that are only partially purified, is highly specific, and serves as a useful screening test for the presence of an antibody or antigen particularly in a heterogeneous sample. However, this method currently is used infrequently in clinical laboratories.
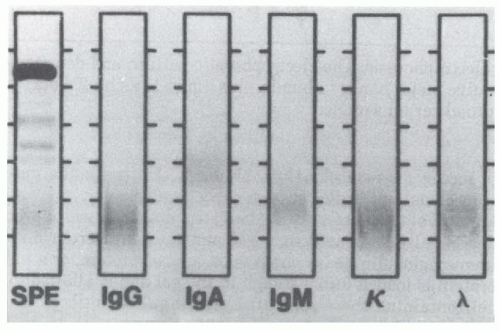 Figure 20-2. Immunofixation electrophoresis. Demonstrates a normal pattern with antibodies to the major heavy-chain and light-chain proteins. A clonal immunoglobulin would result in a darker staining band in a heavy-chain and light-chain zone. |
2. Single radial immunodiffusion allows quantification of a protein (antigen) by adding serum (or other fluid) to wells cut into agarose that contains a specific antiserum. An immunoprecipitin ring of immune complexes is formed as the relevant serum protein (antigen) diffuses into the antibody containing agarose and precipitates once it reaches the region of antibody-antigen equivalence. Thus, the diameter of this ring is proportional to the concentration of the protein (antigen) being evaluated because the concentration of the antiserum is constant throughout the gel. The concentration of the unknown is then determined by plotting the precipitin ring diameter on a concentration curve produced from a series of standards with known protein (antigen) concentration. Radial immunodiffusion (RID) is a simple and reliable method to quantify immunoglobulins (including IgG subclasses), complement components (e.g., C3, C4, and factor B), and other proteins. At least three situations may result in erroneous results using conventional RID kits: (i) low-molecular-weight or monomeric IgM (e.g., Waldenström’s macroglobulinemia and ataxia telangiectasia) can be reported incorrectly high due to the more rapid diffusion of the low-molecular-weight IgM compared with the heavier pentameric IgM standard; (ii) high concentrations of IgG rheumatoid factor can produce complexes of IgG that diffuse more slowly than the IgG standard, resulting in an underestimation of the IgG level; and (iii) the presence of antibovida (e.g., goat) species-directed antibodies, which may be seen in IgA-deficient patients, that can bind the antiserum (in the agarose), if it is from a Bovidae source, yielding a false-positive result.
3. Nephelometry is a method to quantify proteins in a solution that is based on the scattering of light from soluble immune complexes generated by the addition of specific antibody to the sample being tested. In contrast to precipitin reactions, nephelometry is performed in slight antibody excess, and this procedure is readily amenable to automated instrumentation. There are two general approaches, rate and fixed-time nephelometry, both of which enable accurate measurement of IgG and IgG subclasses, IgA, IgM, C3, C4, factor B, C-reactive protein (CRP), and a number of other serum proteins. This method is adaptable for quantifying low-level proteins, including those in the CSF. Nephelometry is the standard method for quantifying immunoglobulin in most clinical laboratories because of the high-volume capabilities of modern nephelometers.
C. Quantitative Assessment (Immunometric Methodology)
1. The radioimmunoassay (RIA) was originally developed to measure insulin levels in serum. The original RIA was based on a competitive binding assay in which a constant amount of antigen-specific antibody was placed together with a small amount of radiolabeled antigen. Next, the sample, containing an unknown concentration of the antigen, was added. Antibody-binding antigen derived from the sample displaces the labeled antigen, and the decrease in radioactivity (in the immune anti-body-antigen complexes) correlated with the concentration of the antigen in the sample.
The competitive binding test can also be reversed by using a fixed concentration of purified antigen together with labeled antibody to
evaluate for specific antibody concentrations in an unknown sample. A further modification of this method uses a solid phase to immobilize one of the reactants (antigen or antibody) and can be performed as an indirect or sandwich assay (see enzyme-linked immunosorbent assay [ELISA] below). RIA is a very sensitive method for specific protein quantification but has the disadvantage of requiring expensive equipment and relatively expensive radioactive reagents, which have defined and often limited shelf lives in addition to requiring special means for disposal. 2. Enzyme-linked immunosorbent assay (ELISA) is an immunoassay method that uses polystyrene plates, tubes, or beads as the solid phase to provide a binding site for the specific antigen under study. Serum (or other fluid) is added to the antigen-coated plate (tube, beads), which enables antibody to bind to the specific antigen immobilized on the solid phase. The presence of bound immunoglobulin from the sample is then detected following a wash step by using a second, enzyme-labeled antibody (reactive with the bound immunoglobulin). Following another wash step, the appropriate chromogenic substrate is added, resulting in enzyme-dependent generation of color (Fig. 20-3). The intensity of this reaction can be measured and is proportional to the antibody concentration present in the sample, derived from a so-called standard curve. The method can be altered using a “sandwich” technique to detect antigen with a test system that has antigen-specific antibody immobilized on the solid phase. This “capture” antibody is used to bind antigen in an unknown sample. This is followed by a second antigen-specific antibody that is enzyme-labeled (Fig. 20-3). Addition of the appropriate chromogenic substrate will yield color that is proportional to the antigen concentration.
evaluate for specific antibody concentrations in an unknown sample. A further modification of this method uses a solid phase to immobilize one of the reactants (antigen or antibody) and can be performed as an indirect or sandwich assay (see enzyme-linked immunosorbent assay [ELISA] below). RIA is a very sensitive method for specific protein quantification but has the disadvantage of requiring expensive equipment and relatively expensive radioactive reagents, which have defined and often limited shelf lives in addition to requiring special means for disposal. 2. Enzyme-linked immunosorbent assay (ELISA) is an immunoassay method that uses polystyrene plates, tubes, or beads as the solid phase to provide a binding site for the specific antigen under study. Serum (or other fluid) is added to the antigen-coated plate (tube, beads), which enables antibody to bind to the specific antigen immobilized on the solid phase. The presence of bound immunoglobulin from the sample is then detected following a wash step by using a second, enzyme-labeled antibody (reactive with the bound immunoglobulin). Following another wash step, the appropriate chromogenic substrate is added, resulting in enzyme-dependent generation of color (Fig. 20-3). The intensity of this reaction can be measured and is proportional to the antibody concentration present in the sample, derived from a so-called standard curve. The method can be altered using a “sandwich” technique to detect antigen with a test system that has antigen-specific antibody immobilized on the solid phase. This “capture” antibody is used to bind antigen in an unknown sample. This is followed by a second antigen-specific antibody that is enzyme-labeled (Fig. 20-3). Addition of the appropriate chromogenic substrate will yield color that is proportional to the antigen concentration.
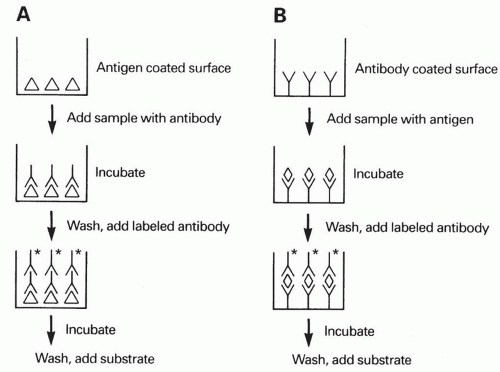 Figure 20-3. Solid-phase enzyme-linked immunosorbent assay (ELISA): (A) indirect assay; (B) “sandwich” assay. The label (*) on the antibody can be an enzyme plus substrate (as shown), a radionuclide (solid-phase radioimmunoassay), or a fluorochrome (solid-phase fluorescence immunoassay). |
The sensitivity of an ELISA can be made comparable to that of an RIA. This method can be used for quantifying a number of specific antibodies or antigens and has the advantages of being simple to perform, requiring no radioactive isotopes, and having excellent reproducibility. It is currently the standard assay in many laboratories for antiviral antibody testing, including initial human immunodeficiency virus (HIV) antibody testing, and also testing for a number of other immunologic proteins (including IgG subclasses, IgE, autoantibodies). Alternatives to the enzyme-based test system include immunoassays that employ either a fluorescent or a chemiluminescent detection system. The latter approach has the advantage of providing amplification to the signal, extending the detection range of the immunoassay.
D. Other Assays for Protein Evaluation
1. Immunoblot (Western blot) is primarily a qualitative method to identify specific antibodies. In this technique, antigens of interest are separated using polyacrylamide gel electrophoresis. The separated proteins are then transferred to a nitrocellulose membrane. This is followed by overlaying the membrane with the antibody (e.g., serum) sample of interest and evaluating for the presence of bound immunoglobulin (specific antibodies) using a second labeled antibody (enzyme or fluorochrome) to human immunoglobulin. Although this method is not quantitative, it does allow for the detection of antibodies reactive with electrophoretically distinct protein antigens. The availability of commercial test kits has simplified the performance of these assays and has allowed the widespread use of the Western blot as a confirmatory test in assessing HIV serologic status and by many laboratories in characterizing the antibody response to Borrelia burgdorferi (Lyme disease). Other clinical laboratory applications of this technology are more limited at this time.
2. Indirect immunofluorescence assays usually use a tissue section or cell line on a microscope slide as a source of the antigen(s) of interest. The substrate is overlaid with the unknown sample, allowing any specific antibody to bind to the antigen(s). The presence of bound antibody is determined with a fluorochrome-labeled antibody reactive with bound antibody, followed by visual examination using fluorescence microscopy. This method is commonly used to test for antinuclear antibody (ANA) and a variety of virus-specific antibodies. The technique is fairly sensitive, relatively quantitative, and technically simple to perform, although reading consistency between different operators can be problematic.
3. Agglutination assays typically are performed using either red blood cells or latex particles that are coated with the antigen of interest. The presence of antigen-specific antibody results in macroscopically detectable agglutination of the antigen-coated particles. These tests were previously used to detect antithyroglobulin and antimicrosomal antibodies (by hemagglutination), rheumatoid factor (by latex agglutination), and various other antibodies. However, this method is not commonly used in the clinical laboratory because it is less sensitive than available immunoassays.
E. New and Emerging Assays
1. Multiplex assays. The development of scaled-down (“miniaturized”) assays that are able to detect and measure multiple analytes simultaneously (e.g., antibodies, cytokines, etc.) has facilitated the study and management
of human diseases and is best represented by multiplex bead array assays (MBAAs). A wide repertoire of tests, using both immunologic and molecular capture targets (ligands), has been developed for MBAA. These include, but are not limited to, determining allergen-specific IgE, infectious agents, antibodies, autoantibodies, cytokines, growth factors, and hormones, as well as a variety of molecular targets of interest (including genes and expression levels of genes). Commercially available MBAA systems may be performed on either multiuse flow cytometers or on more specialized platforms such as the Luminex system. The general principle of the MBAA, with regard to the “multiplexing” capabilities, is achieved through several related approaches. Differential intensities of a single fluorochrome (“color”) or dual fluorochromes contained within the capture bead provide many unique combinations that determine the degree of multiplexing. Dual colors offer more targets/analytes to be measured but may need more sophisticated platforms, while single fluorochrome can often be adapted for a clinical cytometer that is already in use.
of human diseases and is best represented by multiplex bead array assays (MBAAs). A wide repertoire of tests, using both immunologic and molecular capture targets (ligands), has been developed for MBAA. These include, but are not limited to, determining allergen-specific IgE, infectious agents, antibodies, autoantibodies, cytokines, growth factors, and hormones, as well as a variety of molecular targets of interest (including genes and expression levels of genes). Commercially available MBAA systems may be performed on either multiuse flow cytometers or on more specialized platforms such as the Luminex system. The general principle of the MBAA, with regard to the “multiplexing” capabilities, is achieved through several related approaches. Differential intensities of a single fluorochrome (“color”) or dual fluorochromes contained within the capture bead provide many unique combinations that determine the degree of multiplexing. Dual colors offer more targets/analytes to be measured but may need more sophisticated platforms, while single fluorochrome can often be adapted for a clinical cytometer that is already in use.
The beads with their unique spectral color (property, “address”) are coated (tagged, immobilized) with an antigen/ligand that forms the target for detecting the analyte (such as an antibody or cytokine) in the serum/plasma (only small amounts of serum/plasma are typically required) of the individual being tested. The reaction is developed by subsequent detection of the captured ligand using a second “reporter” antibody in a high throughput and largely automated manner with sophisticated data processing and analysis.
MBAA technology differs from ELISA technology in the following ways: ELISAs do not use fluorescence as the read-out approach and, generally, measure only one analyte at a time (i.e., uniplex). As ELISA has been the prevailing (preceding) method for many analytes, it is necessary to compare both methodologies. For example, a key concern in the development of a clinically applicable MBAA is the possibility that the process of multiplexing itself introduces artifacts in the assay (“matrix” effect). Commercial kits are generally optimized to eliminate or minimize matrix effects (requiring strict adherence to the manufacturer’s protocols).
The correlations between multiplex (MBAA) and corresponding uniplex (ELISA) measurements are generally good, especially when identical capture and reporter substrates (antibodies), as well as similar reagents (e.g., diluents, serum blockers, etc.), are used. When developing MBAA assays for diagnosis or sequential monitoring of patients, the MBAA should first be compared to the existing methodology, and then use a consistent MBAA approach (kit, instrumentation, reagents).
2. Luciferase immunoprecipitation systems (LIPSs). Among the challenges faced when assessing antigen-specific antibodies is the need to know the nature of antigens and the generation of antibodies that are specific for those antigens. With infectious agents, it is often important to distinguish the pathogen-specific antigen(s) or epitope(s) that are relevant for protective immunity from the potentially vast array of epitopes/antigenic structures that are not. In addition, accurate detection of an antibody response to an infectious agent (“seroconversion”) may depend on the conformational nature of the antigen(s), rather than strictly linear epitopes. Obtaining the antigens of interest from whole
cell lysates of infectious agents or through recombinant technology may lead to cross-reactivity, high background activation or potentially, the use of antigens or antigenic structures that are not relevant in vivo.
cell lysates of infectious agents or through recombinant technology may lead to cross-reactivity, high background activation or potentially, the use of antigens or antigenic structures that are not relevant in vivo.
The LIPSs assay is an emerging technology that can be used to address these challenges. With LIPSs, the combination of synthetic biology involving the engineering of proteins of interest and a novel immunoprecipitation technology allows antigen and antibody to interact with each other in solution, rather than bound to plates (solid phase), so that in vivo conformational relevance of the interaction can be mimicked in vitro.
On the synthetic biology side, artificial gene synthesis can generate multiepitope and chimeric proteins that better simulate how an antigenic structure, for example, derived from a pathogen-associated antigen, interacts with antibody in vivo. The proteins are generated using mammalian expression vectors. Advances in proteomics will expand the ability to generate antigens/proteins that can be used to detect antibodies in biologic samples. In order to develop an immunoassay, the proteins are fused with a luminescent enzyme (Renilla luciferase) that produces a light signal when exposed to its substrate (e.g., coelenterazine).
The combination of selecting mammalian cells to generate recombinant proteins and allowing the proteins and (patient-derived) antibodies to interact in solution enhances the sensitivity of the assay with dynamic ranges that are substantially higher than that obtained by traditional ELISA technology. Similarly, the same technology can be used to develop immunoassays that can detect and measure autoantibodies and tumor-associated antibodies.
II. EVALUATION OF LYMPHOCYTES
A. Characterizing Lymphocytes
Flow cytometry provides a methodology for multiparameter analysis of blood cells at the single-cell level. It is the most versatile platform to study the human immune system and is commonly used for the identification of human lymphocyte subsets, a method referred to as immunophenotyping. Immunophenotypic studies provide insights into the differentiation and function of human lymphocytes and identification of novel lymphocyte subsets, such as natural killer T cells (NK T cells), Th17 T cells, among many others. An (ever) increasing repertoire of monoclonal antibodies, specific for lymphocyte-specific antigens, allows for continually increasing the types of cells that can be evaluated. Along with advances in instrument operation, automation, data analysis, and fluorochrome chemistry, multiparameter analysis (up to 15 or more parameters per individual cell) is possible.
B. Principles of Flow Cytometry
1. Monoclonal antibodies are produced by fusing an immortal murine myeloma cell line with normal murine plasma cells to yield hybridoma cells that effectively are immortal and can be selected for appropriate antibody specificity before cell expansion. This approach leads virtually to an unlimited supply of antigen-specific antibodies. There is an everincreasing number of monoclonal antibodies to lymphocyte cell surface antigens, which are categorized according to a cluster of differentiation
(CD) numerical convention (Table 20-1, http://en.wikipedia.org/wiki/List_of_human_clusters_of_differentiation). These reagents allow for significant progress in the understanding of lymphocyte differentiation and maturation. In addition, other monoclonal antibodies yield information about mechanisms of cell adhesion and activation. In the area of lymphocyte phenotyping, monoclonal antibodies enable more complete evaluation of cell lineage, differentiation, activation, and functional capacity. These reagents provide important pieces of information regarding the presence of particular cells and their potential function, but they do not evaluate the actual function of the cell. More recently, intracellular staining for specific proteins including cytokines has become a standard approach that supplements immunophenotyping.
(CD) numerical convention (Table 20-1, http://en.wikipedia.org/wiki/List_of_human_clusters_of_differentiation). These reagents allow for significant progress in the understanding of lymphocyte differentiation and maturation. In addition, other monoclonal antibodies yield information about mechanisms of cell adhesion and activation. In the area of lymphocyte phenotyping, monoclonal antibodies enable more complete evaluation of cell lineage, differentiation, activation, and functional capacity. These reagents provide important pieces of information regarding the presence of particular cells and their potential function, but they do not evaluate the actual function of the cell. More recently, intracellular staining for specific proteins including cytokines has become a standard approach that supplements immunophenotyping.
2. Fluorochromes are compounds that absorb light of a defined wavelength and convert this energy into light of a longer defined wavelength (lower energy). There are currently a wide range of fluorochromes routinely used in clinical phenotyping including fluorescein isothiocyanate (FITC), phycoerythrin (PE), and peridinin chlorophyll protein (PerCP) that are excited by blue light (488 nm), with FITC generating green, PE emitting orange, and PerCP producing red light. Additionally, allophycocyanin and double (tandem) fluorochromes, which rely on energy transfer from one component to the other, are exciteable by blue and red light lasers, which now come standard in most benchtop flow cytometers, extending the number of colors available for clinical use. Other fluorochromes, including amcyan, pacific blue, and quantum dot nanocrystals, are used primarily in research settings because they require a violet or ultraviolet excitation source or specialized filter sets.
3. The flow cytometer




Related posts:



Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
