Introduction
In this chapter we attempt to provide a useful guide to the rapid and accurate diagnosis of anemia in children and young adults. We do not dwell on each diagnosis because they are described in detail throughout the book.
Though productive methods of differential diagnosis of anemia vary greatly with the age of the pediatric patient, the most valuable tools at all ages remain the history and physical examination. Even in the neonate the history of the pregnancy, the family history, and the events surrounding birth are paramount, and the appearance of the newborn baby strongly influences diagnosis. In fact, the history and physical examination may well be more useful than examination of the blood because the morphology of the red cells of normal neonates may be strikingly abnormal. Morphology of neonatal blood can be very deceiving. As mentioned below, anemia is due to either hemorrhage, failure of erythropoiesis, accelerated hemolysis, or combinations of the three. The symptoms of anemia are well established. They are due either to sudden blood volume loss or to chronic redistribution of a low blood volume with a high fraction sequestered in the central blood volume, both complicated by low oxygen carrying capacity. The results are weakness, dyspnea and fatigue, cold intolerance, dizziness, and depression. If the blood volume loss is substantial and sudden, the patient may present in a state of collapse. If anemia develops slowly, low oxygen carrying capacity may be compensated by a higher cardiac output due to the rise in central blood volume and a lowering of blood viscosity and by increased erythrocyte 2,3-DPG. In fact, patients with slowly developing anemia to as low as 1 g of hemoglobin (Hb) per 100 mL of blood have been observed to walk into clinics. But this is surely a feat of the young. Older patients with less accommodating vascular trees are not capable of such gymnastics. Woe to such a patient who encounters a physician determined to transfuse with red cells without removing an equal volume of watery blood. The maneuver will surely drive the victim’s massively expanded central blood volume into pulmonary edema.
The rate of development of symptoms provides strong etiologic clues because the normal red cell life span is 3.5 months, and cardiac compensation for a falling oxygen carrying capacity can blunt symptoms. Thus a patient who suddenly complains of severe fatigue and is noticeably pale has probably lost red cells rapidly. This must be due to hemolysis or bleeding. If the symptoms accumulate slowly, this is likely due to failure of erythropoiesis that may be complicated by malnutrition, endocrine deficiencies such as thyroid or adrenal failure, and/or by renal insufficiency or inflammatory disease with concomitant inhibition of iron absorption.
The physical examination of the anemic patient may be particularly revealing of etiology. Pallor is nonspecific, but when tinged with jaundice, it may suggest increased hemolysis. This is readily confirmed if the foam of shaken urine remains white. Yellow skin or sclera without biliuria strongly indicates that the circulating bilirubin has NOT been conjugated and is the direct product of rapidly hemolyzed red cells. Indeed, in the absence of liver disease, the level of unconjugated bilirubin in the peripheral blood is a direct function of red cell turnover. The relationship is confusing in the neonate because of its physiologic defect in the conjugation pathway and is not accurate in liver disease.
Examination of the tongue and oral mucosa will reveal vitamin, iron, and corticosteroid deficiencies, while stature and the skeleton, particularly the forehead, face, and skull, may indicate the chronicity of hemolysis and concomitant energy consumption and marrow space expansion. The digits may reveal boney abnormalities seen in congenital anemias. Enlarged lymph nodes and their texture, together with hepatosplenomegaly, may suggest invasive or infectious diseases, as do petechiae or other skin lesions. The pulse rate and blood pressure and the heave of the heart against the chest may reveal the length and severity of anemia, while the lungs may demonstrate the incipiency of pulmonary edema. A walk up the stairs with comparison of patient and caretaker pulse rates may be highly revealing. Neurologic examination may show the characteristic slow return of Achilles tendon reflexes seen in hypothyroidism or the posterior column defects of vitamin B 12 deficiency.
Definition of Anemia
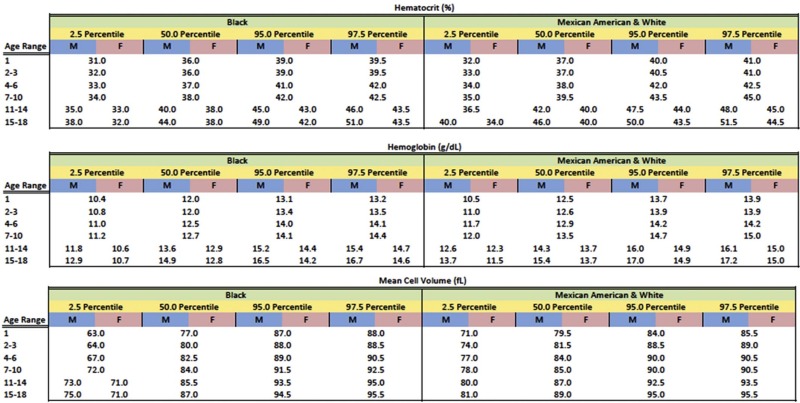
Anemia is generally defined as a reduction in red cell mass or blood Hb concentration. The limit for differentiating anemic from normal states is usually set at 2 standard deviations below the mean for the normal population. This definition will result in 2.5% of the normal population being classified as anemic. Conversely, values for Hb-deficient individuals are distributed in such a fashion that some are placed within the normal range. These individuals, who have the potential for an Hb concentration in the upper part of the normal range, may be recognized only after a response to treatment. Definition of the lower limit of normal for both pediatric and adult patients is critically dependent on the population used to establish the normal range of values. Hematocrit (Hct), Hb, and mean corpuscular volume (MCV) are significantly lower in African Americans than in Caucasians. A third of this difference can be accounted for by the α-globin 3.7 kb deletion. In the absence of a race-specific normal range, the fact that African Americans generally have lower Hb levels than Caucasians (approximately 0.5 g/dL) could result in 10% of normal African-American children being considered anemic just because the proper normal range is not used. Figure 9-1 presents pediatric reference ranges for Hb in Caucasians, African Americans, and Mexican Americans that were obtained from the National Health and Nutrition Examination Survey (NHANES) III database.
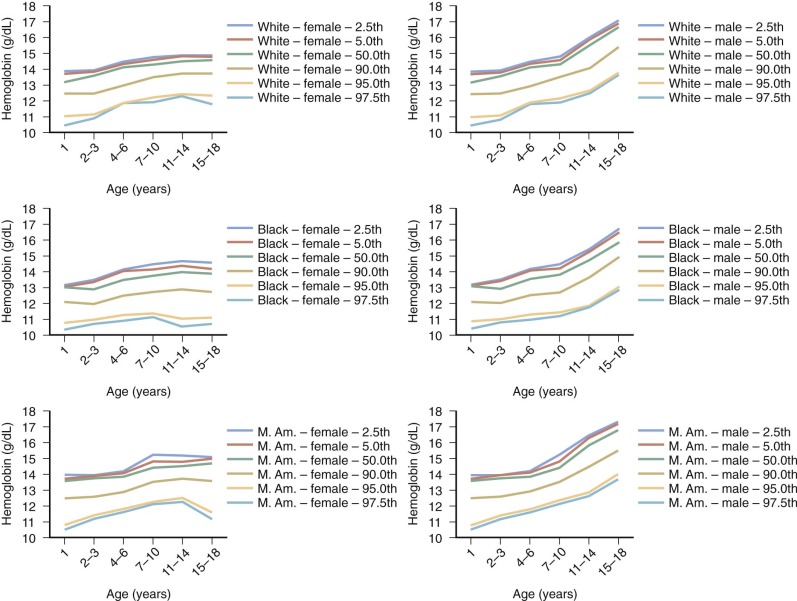
Because the primary function of the red cell is to deliver and release adequate quantities of oxygen to tissues to meet their metabolic demands, it is apparent that some measures of both body oxygen metabolism and accompanying cardiovascular compensation are required to complement the current laboratory definition of anemia. The fact that Hb concentration alone is insufficient to judge whether a patient is “functionally anemic” is best illustrated in a patient with cyanotic congenital heart disease or chronic respiratory insufficiency or in one with mutant Hbs that alter Hb’s affinity for oxygen (see Chapter 19 ).
With these caveats in mind, a useful statistical definition of anemia that recognizes the effect of age, gender, and ethnicity on the designation of anemia is provided in Figure 9-2 .
Classification of Anemias
Anemias may be classified on the basis of physiology or morphology. A combination of both approaches is often used in the initial differential diagnosis.
To determine and distinguish the multiple disorders capable of producing anemia, one must separate the causes of anemia into two categories of functional disturbances:
- 1.
Disorders of effective red cell production, in which the net rate of red cell production is depressed. This can be due to disorders of erythrocyte maturation, in which erythropoiesis is present but ineffective, or to an absolute failure of erythropoiesis. In the former the bone marrow may contain many erythroblasts that die in situ before reaching the reticulocyte stage. In the latter there is absolute erythroblastopenia.
- 2.
Disorders in which rapid erythrocyte destruction or red cell loss is primarily responsible for the anemia.
These two categories are not mutually exclusive. More than one mechanism may be present in some anemias, but one functional disorder is generally the major reason for the patient’s anemia. Box 9-1 lists the anemias most commonly encountered in infancy and childhood and classifies them into three categories of functional disturbance.
- 1.
Disorders of red cell production in which the rate of red cell production is less than expected for the degree of anemia
- a.
Marrow failure
- •
Aplastic anemia
Congenital
Acquired
- •
Pure red cell aplasia
Congenital
Diamond-Blackfan syndrome
Aase’s syndrome
Acquired
Transient erythroblastopenia of childhood
Human parvovirus B19 infection
- •
Marrow replacement
Malignancies
Osteopetrosis
Myelofibrosis
Chronic renal disease
Vitamin D deficiency
Granulomatous infection
- •
Pancreatic insufficiency–marrow hypoplasia syndrome
- •
Fanconi’s anemia
- •
- b.
Impaired erythropoietin production
- •
Chronic renal disease
- •
Hypothyroidism, hypopituitarism
- •
Chronic inflammation
- •
Protein malnutrition
- •
Hemoglobin mutants with decreased affinity for oxygen
- •
- c.
Anemia of chronic disease
- a.
- 2.
Disorders of erythroid maturation and ineffective erythropoiesis
- a.
Abnormalities in cytoplasmic maturation
- •
Iron deficiency
- •
Thalassemia syndromes
- •
Sideroblastic anemias
- •
Lead poisoning
- •
- b.
Abnormalities in nuclear maturation
- •
Vitamin B 12 deficiency
- •
Folic acid deficiency
- •
Thiamine-responsive megaloblastic anemia
- •
Hereditary abnormalities in folate metabolism
- •
Orotic aciduria
- •
Zinc-induced copper deficiency
- •
- c.
Congenital dyserythropoietic anemias (types I, II, III, IV)
- d.
Erythropoietic protoporphyria
- e.
Refractory sideroblastic anemia with vacuolization of marrow precursors and pancreatic dysfunction/deficiency
- a.
- 3.
Hemolytic anemias
- a.
Defects in hemoglobin
- •
Structural mutants
- •
Synthetic mutants (thalassemia syndromes)
- •
- b.
Defects in the red cell membrane
- c.
Defects in red cell metabolism
- d.
Antibody mediated
- e.
Mechanical injury to the erythrocyte
- f.
Thermal injury to the erythrocyte
- g.
Oxidant-induced red cell injury
- h.
Infectious agent–induced red cell injury
- i.
Paroxysmal nocturnal hemoglobinuria
- j.
Plasma lipid–induced abnormalities in the red cell membrane
- a.
Anemias may also be classified on the basis of red cell size and then further subdivided according to red cell morphology. In this type of classification, anemias are subdivided into microcytic, normocytic, and macrocytic anemias. This classification is also arbitrary, and categories are not mutually exclusive. For example macrocytic reticulocytes abound in the hemolytic anemias. Therefore, although mature erythrocytes in the various hemolytic anemias may be normocytic, the MCV of all the cells may be larger than normal because of the contribution of reticulocytes to the volume measurement. Volume distribution curves may reveal the contribution of a subset of large cells to the MCV. Furthermore, during the course of a disease, classification of the patient’s anemia may change from one category to another as a result of other clinical or pathologic variables. In Box 9-2 the more common anemias of infancy and childhood are classified on the basis of their characteristic cell size.
Microcytic anemias
Iron deficiency (nutritional, chronic blood loss)
Chronic lead poisoning
Thalassemia syndromes
Sideroblastic anemias
Chronic inflammation
Some congenital hemolytic anemias with unstable hemoglobin
Macrocytic Anemias
With megaloblastic bone marrow
- •
Vitamin B 12 deficiency
- •
Folic acid deficiency
- •
Hereditary orotic aciduria
- •
Thiamine-responsive anemia
- •
Without megaloblastic bone marrow
- •
Aplastic anemia
- •
Diamond-Blackfan syndrome
- •
Hypothyroidism
- •
Liver disease
- •
Bone marrow infiltration
- •
Dyserythropoietic anemias
- •
Normocytic Anemias
Congenital hemolytic anemias
- •
Hemoglobin mutants
- •
Red cell enzyme defects
- •
Disorders of the red cell membrane
- •
Acquired hemolytic anemias
- •
Antibody mediated
- •
Microangiopathic hemolytic anemias
- •
Secondary to acute infections
- •
Acute blood loss
Splenic pooling
Chronic renal disease (usually)
Evaluation of the Anemic Patient
Initial Encounter
The initial diagnostic approach to an anemic patient includes a detailed history and physical examination and a minimum of essential laboratory tests. Box 9-3 and Table 9-1 list the features of the history and physical examination that are most helpful in providing clues to the cause of the anemia. Often they may be more diagnostic than the laboratory studies.
Family History: Because the prevalence of hemoglobinopathies and thalassemias varies greatly among different ethnic backgrounds, it is important to inquire about ethnicity. A family history of anemias or symptoms that may be related to anemias, like jaundice, gallstones, cholecystectomy/splenectomy, and transfusion, in family members should be investigated. A family history of blood loss (menorrhagia, epistaxis) may suggest that the anemia is due to the presence of an inherited bleeding disorder.
Personal History: With almost universal newborn screening in the United States and other developed countries, many patient encounters are generated by the identification of a hemoglobinopathy by newborn screening in the absence of any symptomatology. For all other circumstances, the history should focus on the characterization of possible hemolytic crises or episodes of blood loss. Menstrual history should be obtained in all girls. If the patient has recently moved from or traveled to an underdeveloped country, intestinal parasite infestation (hookworm, whipworm) should be considered, as well as malaria and infectious hepatitis. A dietary history is crucial to identify conditions favoring folate deficiency or iron deficiency. A detailed medication history is of great importance to identify drugs that may directly produce anemia or may be contributing to anemia via increased blood losses (e.g., aspirin, nonsteroidal antiinflammatory drugs).
Physical Examination: The presence of congenital defects is an important feature of congenital anemias, such as Fanconi’s anemia (FA). Skeletal abnormalities (hypoplastic or absent thumb/radius) are present in at least half of patients with FA. Many of these patients also have short stature.
Age
Nutritional iron deficiency is never responsible for anemia in term infants before 6 months of age and rarely seen in premature infants before the time that they have doubled their birth weight. Anemia occurring in the neonatal period is generally the result of recent blood loss, isoimmunization, or initial manifestation of a congenital hemolytic anemia or congenital infection. Anemia first detected at 3 to 6 months of age suggests a congenital disorder of hemoglobin synthesis or hemoglobin structure.
Gender
Consider X-linked disorders in males (G6PD deficiency, pyruvate kinase deficiency).
Race
Hemoglobins S and C more common in African Americans; β-thalassemia more common in Caucasians; α-thalassemia trait most common among African Americans and Asians
Ethnicity
Thalassemia syndromes most common among patients of Mediterranean origin. G6PD deficiency observed more often among Sephardic Jews, Filipinos, Greeks, Sardinians, and Kurds
Neonatal
A history of hyperbilirubinemia in the newborn period suggests the presence of congenital hemolytic anemia, such as the hereditary spherocytosis of G6PD deficiency. Prematurity predisposes to the early development of iron deficiency.
Diet
Document sources of iron, vitamin B 12 , folic acid, or vitamin E in the diet. A history of pica, geophagia, or pagophagia suggests the presence of iron deficiency.
Drugs
Oxidant-induced hemolytic anemia, phenytoin (Dilantin)-induced megaloblastic anemia, drug-induced aplastic anemia
Infection
Hepatitis-induced aplastic anemia, infection-induced red cell aplasia, hemolytic anemia
Inheritance
Family history of anemia, jaundice, gallstones, or splenomegaly
Diarrhea
Suspect small bowel disease with malabsorption of folate or vitamin B 12 . Suspect inflammatory bowel disease with blood loss. Suspect protein-losing enteropathy with blood loss.
G6PD, Glucose-6-phosphate dehydrogenase.
| Finding | Cause | |
|---|---|---|
| Skin | Hyperpigmentation | Fanconi’s aplastic anemia |
| Petechiae, purpura | Autoimmune hemolytic anemia with thrombocytopenia, hemolytic-uremic syndrome, bone marrow aplasia, bone marrow infiltration | |
| Carotenemia | Suspect iron deficiency in infants | |
| Jaundice | Hemolytic anemia, hepatitis, aplastic anemia | |
| Cavernous hemangioma | Microangiopathic hemolytic anemia | |
| Ulcers on lower extremities | S and C hemoglobinopathies, thalassemia | |
| Facies | Frontal bossing, prominence of the malar and maxillary bones | Congenital hemolytic anemias, thalassemia major, severe iron deficiency |
| Eyes | Microcornea | Fanconi’s aplastic anemia |
| Tortuosity of the conjunctival and retinal vessels | S and C hemoglobinopathies | |
| Microaneurysms of the retinal vessels | S and C hemoglobinopathies | |
| Cataracts | Glucose-6-phosphate dehydrogenase deficiency, galactosemia with hemolytic anemia in the newborn period | |
| Vitreous hemorrhages | S hemoglobinopathy | |
| Retinal hemorrhages | Chronic, severe anemia | |
| Edema of the eyelids | Infectious mononucleosis, exudative enteropathy with iron deficiency, renal failure | |
| Blindness | Osteopetrosis | |
| Mouth | Glossitis | Vitamin B 12 deficiency, iron deficiency |
| Angular stomatitis | Iron deficiency | |
| Chest | Unilateral absence of the pectoral muscles | Poland’s syndrome (increased incidence of leukemia) |
| Shield chest | Diamond-Blackfan syndrome | |
| Hands | Triphalangeal thumbs | Red cell aplasia |
| Hypoplasia of the thenar eminence | Fanconi’s aplastic anemia | |
| Spoon nails | Iron defiency | |
| Spleen | Enlargement | Congenital hemolytic anemia, leukemia, lymphoma, acute infection, portal hypertension |
There is substantial variability in the effectiveness of the compensatory mechanisms for anemia: While in some cases the severity of the anemia is evident based on the presence of tachycardia, lethargy, or pallor, in other cases a very severe, chronic anemia may be associated with a paucity of symptoms. Short stature and skeletal changes induced by expansion of the erythroid mass should also be noted because they are often the result of anemia and expansion of erythroid marrow.
Selection of Appropriate Laboratory Tests ( Box 9-4 )
First-Tier Tests
- •
The initial laboratory tests should always include a complete blood count (CBC) with white blood cell differential, a reticulocyte count, and an examination of a peripheral blood smear. If a test for the identification of Hb variants is included in this initial panel, high-performance liquid chromatography, capillary electrophoresis, or isoelectrofocusing should be preferred over classic cellulose acetate and acid-citrate Hb electrophoresis. A glucose-6-phosphate dehydrogenase screening test, total and direct bilirubin, lactate dehydrogenase (LDH), and iron studies (serum iron, total iron binding capacity, and ferritin concentrations) should also be considered as part of the initial laboratory evaluation of the anemic child. It should be noted that LDH is not a marker of hemolysis: Elevated LDH is an indication of intravascular hemolysis. With reliable clinical grade assays now in place, serum hepcidin might be included if iron deficiency is unexplained.
- •
Second-Tier Tests
- •
Based on the initial evaluation of the patient and the first round of laboratory tests, additional disease-specific tests should be considered. In the presence of macrocytosis, plasma B 12 , folate, and perhaps homocystein and serum/urine methylmalonic acid should be determined together with a measurement of thyroid hormone function. Osmotic fragility (nonincubated and incubated) should be considered in the presence of possible hereditary spherocytosis (HS)/xerocytosis, although the presence of cells with high mean corpuscular Hb concentration (MCHC) is usually diagnostic of HS in the absence of hemoglobinopathy-induced cation loss, antibody-induced hemolysis, hereditary xerocytosis, and transient-hypoproliferative states. Serum haptoglobin, serum/plasma Hb, and urine hemosiderin should be considered in working up possible chronic hemolytic conditions as well as a direct and indirect antiglobulin test or Coombs test and red-cell or white-cell based flow cytometric assays for paroxysmal nocturnal hemoglobinuria (PNH).
- •
Supravital staining of erythrocytes (crystal violet to detect Heinz bodies or denatured Hb, brilliant cresyl blue for unstable Hb), a screening test for the presence of unstable hemoglobins like heat or isopropanol stability, should be considered if there are indications of possible unstable hemoglobinopathy or oxidant-induced hemolysis.
- •
Erythrocyte protoporphyrin is of limited value in the diagnosis of iron deficiency or lead poisoning. However, increased levels of erythrocyte protoporphyrin help to distiguish X-linked sideroblastic anemia type A (XLSA/A) due to deficiency in either ABCB7, SLC19A2, or other mitochondrial defects from classic XLSA due to ALAS2 mutations, which characteristically has low or normal levels.
- •
Erythrocyte adenosine deaminase, serum α-fetoprotein, mitomycin, or diepoxybutane tests are indicated for Diamond-Blackfan anemia (DBA) and FA, respectively.
- •
Family studies are helpful in the presence of challenging combinations of hemoglobinopathies/thalassemias and in HS/xerocytosis. Specialized red blood cell (RBC) enzyme studies should be considered with the caveat that only a handful of reference laboratories in the United States offer functional studies for red cell enzymopathies.
- •
To diagnose erythroid suppression induced by human parvovirus B19 infection, immunoglobulin M (IgM) and immunoglobulin G (IgG) serum titers and viral DNA detection tests should be considered.
- •
Serum erythropoietin (EPO) levels are indicated only when there is a question of inappropriately low EPO for the degree of anemia, provided that the laboratory has generated an EPO normal range as a function of Hct/Hb values.
- •
Third-Tier Tests
- •
Bone marrow studies should be performed when there is clear evidence of a hypoproliferative anemia, except for cases of transient virus-induced erthroblastopenia, in which they are not indicated. Bone marrow studies are required to diagnose the presence of ringed sideroblasts, which are a distinguishing sign of sideroblastic anemias, to identify megaloblastic erythropoiesis and when there is a suspicion of an associated hematologic malignancy based on peripheral blood morphology.
- •
DNA-based diagnosis: Ordering of molecular studies should be the result of a proper balancing between the curiosity of the physician and the needs and possible benefits for the patient. Cost of these tests should also be considered, and whether they are associated with significant cost borne by the patient or family or by insurance carriers. For most of red cell membrane and red cell enzyme disorders, molecular testing rarely results in measurable benefits for the patients and their families and for the clinical management of the disease. Genetic testing has a value in congenital dyserythropoietic anemias (CDAs) and in DBA.
- •
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


