Disorder
Disease definition
References
Monoclonal gammopathy of undetermined significance (MGUS)
All 3 criteria must be met:
•Serum monoclonal protein <3 g/dL
•Clonal bone marrow plasma cells <10 %, and
•Absence of end-organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to the plasma cell proliferative disorder; or in the case of IgM MGUS no evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
[3]
Smoldering multiple myeloma (also referred to as asymptomatic multiple myeloma)
Both criteria must be met:
•Serum monoclonal protein (IgG or IgA) ≥3 g/dL and/or clonal bone marrow plasma cells ≥10 %, and
•Absence of end-organ damage such as lytic bone lesions, anemia, hypercalcemia, or renal failure that can be attributed to a plasma cell proliferative disorder
[3]
Multiple myeloma
All three criteria must be met except as noted:
•Clonal bone marrow plasma cells ≥10 % and/or biopsy proven plasmacytoma
•Presence of serum and/or urinary monoclonal protein (except in patients with true non-secretory multiple myeloma), and
•Evidence of end organ damage that can be attributed to the underlying plasma cell proliferative disorder, specifically
Hypercalcemia: Serum calcium > 11.5 mg/dL or
Renal insufficiency: Serum creatinine > 1.73 mmol/L (or >2 mg/dL) or estimated creatinine clearance less than 40 ml/min
Anemia: Normochromic, normocytic with a hemoglobin value of >2 g/dL below the lower limit of normal or a hemoglobin value <10 g/dL
Bone lesions: Lytic lesions, severe osteopenia, or pathologic fractures
IgM Monoclonal gammopathy of undetermined significance (IgM MGUS)
All three criteria must be met:
•Serum IgM monoclonal protein <3 g/dL
•Bone marrow lymphoplasmacytic infiltration <10 %, and
•No evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
Smoldering Waldenström’s macroglobulinemia (also referred to as indolent or asymptomatic Waldenström’s macroglobulinemia)
Both criteria must be met:
•Serum IgM monoclonal protein ≥3 g/dL and/or bone marrow lymphoplasmacytic infiltration ≥10 %, and
•No evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
Waldenström’s macroglobulinemia
All criteria must be met:
•IgM monoclonal gammopathy (regardless of the size of the M protein), and
≥10 % bone marrow lymphoplasmacytic infiltration (usually intertrabecular) by small lymphocytes that exhibit plasmacytoid or plasma cell differentiation and a typical immunophenotype (e.g., surface IgM+, CD5+/−, CD10−, CD19+, CD20+, CD23− that satisfactorily excludes other lymphoproliferative disorders including chronic lymphocytic leukemia and mantle cell lymphoma
•Evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
Solitary Plasmacytoma
All four criteria must be met
•Biopsy proven solitary lesion of bone or soft tissue with evidence of clonal plasma cells
•Normal bone marrow with no evidence of clonal plasma cells
•Normal skeletal survey and MRI of spine and pelvis (except for the primary solitary lesion)
•Absence of end-organ damage such as hypercalcemia, renal insufficiency, anemia, or bone lesions (CRAB) that can be attributed to a lympho-plasma cell proliferative disorder
Systemic AL amyloidosis
All four criteria must be met:
•Presence of an amyloid-related systemic syndrome (such as renal, liver, heart, gastrointestinal tract, or peripheral nerve involvement)
•Positive amyloid staining by Congo red in any tissue (e.g., fat aspirate, bone marrow, or organ biopsy)
•Evidence that amyloid is light-chain related established by direct examination of the amyloid (possibly using Mass Spectrometry (MS)-based proteomic analysis, or immuno-electronmicroscopy; note that immunohistochemistry results to type amyloid may be unreliable), and
•Evidence of a monoclonal plasma cell proliferative disorder (serum or urine M protein, abnormal free light chain ratio, or clonal plasma cells in the bone marrow)
Note: Approximately 2–3 % of the patients with AL amyloidosis will not meet the requirement for evidence of a monoclonal plasma cell disorder listed previously; the diagnosis of AL amyloidosis must be made with caution in these patients
[12]
POEMS syndrome
All four criteria must be met:
•Presence of a monoclonal plasma cell disorder
•Peripheral neuropathy
•Osteosclerotic bone lesions or Castleman’s disease, and
•At least one of the following features: Elevations in plasma or serum levels of vascular endothelial growth factor, organomegaly, endocrinopathy (excluding diabetes mellitus or hypothyroidism), edema, typical skin changes, papilledema, thrombocytosis, or polycythemia
Note: Not every patient meeting the previous criteria will have POEMS syndrome; the features should have a temporal relationship to each other and no other attributable cause. Anemia and/or thrombocytopenia are distinctively unusual in this syndrome unless Castleman disease is present
[13]
Clinical Features
The most common presenting symptoms of multiple myeloma are fatigue and bone pain [17]. Osteolytic bone lesions and/or compression fractures are hallmarks of the disease, which can be detected on routine radiographs, magnetic resonance imaging (MRI), or computed tomographic (CT) scans, and may cause significant morbidity [18]. Bone pain may present as an area of persistent pain or migratory bone pain, often in the lower back and pelvis. Pain may be sudden in onset when associated with a pathological fracture, and is often precipitated by movement. Extramedullary expansion of bone lesions may cause nerve root or spinal cord compression. Anemia occurs in 70 % of the patients at diagnosis and is the primary cause of fatigue. Hypercalcemia is found in 15 % of the patients, while the serum creatinine is elevated in almost one-half [17].
Diagnosis
Disease Definition
The diagnosis of active myeloma requires 10 % or more plasma cells on bone marrow examination (or biopsy proven plasmacytoma), M protein in the serum and/or urine (except in patients with true non-secretory myeloma) and evidence of end-organ damage (hypercalcemia, renal insufficiency, anemia, or bone lesions) secondary to the underlying plasma cell disorder (Table 33.1) [19]. Almost all patients evolve from an asymptomatic pre-malignant stage termed monoclonal gammopathy of undetermined significance (MGUS) [20, 21]. MGUS is present in over 3 % of the population above the age of 50, and progresses to myeloma or related malignancy at a rate of 1 % per year [22, 23]. In some patients, an intermediate asymptomatic but more advanced pre-malignant stage referred to as smoldering multiple myeloma (SMM) can be recognized clinically [24, 25].
Laboratory Testing for Diagnosis
When multiple myeloma is suspected, patients should be tested for the presence of monoclonal (M) proteins using a combination of tests that should include serum protein electrophoresis (SPEP), serum immunofixation (SIFE) (Fig. 33.1a, b), urine protein electrophoresis (UPEP), and urine immunofixation (UIFE). The serum free light chain (FLC) assay can be used for screening in place of the urine studies mentioned earlier [26]. Only 82 % of the patients with myeloma have an M protein that is detectable on SPEP, whereas SIFE will identify an M protein in 93 % of the patients [17]. Up to 20 % of the patients with multiple myeloma lack heavy-chain expression in the M protein, and are considered to have light-chain multiple myeloma. The M protein in these patients is always detected in the urine, and can be absent even on SIFE, making it necessary that urine studies or the serum FLC assay are always done in all patients in whom myeloma is suspected. Combining serum studies with either urine studies or the serum FLC assay will reveal an M protein in 97–98 % of the patients with myeloma [17, 26]. The presence of an M protein is indicative of an underlying clonal plasma cell proliferative disorder, and further testing is required to diagnose myeloma, and to distinguish it among the various plasma cell disorders (Table 33.1). Approximately 3 % of the patients with multiple myeloma have true non-secretory disease and have no evidence of an M protein on any of the studies mentioned earlier [17].
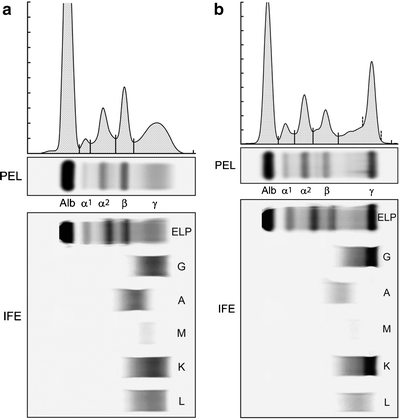
Fig. 33.1
(a) Serum protein electrophoresis (PEL) and immunofixation (IFE) showing a normal pattern with no evidence of monoclonal protein. (b) Serum protein electrophoresis (PEL) showing a monoclonal (M) protein in the gamma region, which is IgG lambda on immunofixation (IFE)
A complete blood count (CBC) with differential, liver function tests, urinalysis, serum electrolytes, including serum creatinine and calcium, beta-2 microglobulin, albumin, C-reactive protein, and lactate dehydrogenase (LDH) is needed for diagnosis, prognosis, and staging. Serum CrossLaps to measure carboxy-terminal collagen crosslinks (CTX) may be useful in assessing bone turnover, and determine the adequacy of bisphosphonate therapy [27, 28].
Bone Marrow Studies
A unilateral bone marrow aspiration and biopsy is indicated in all patients with suspected multiple myeloma. By definition, all patients should have bone marrow involvement with ≥ 10 % clonal plasma cells (or have a biopsy proven plasmacytoma). If a lesser extent of involvement is detected, one is either dealing with an erroneous diagnosis or there is a sampling error due to patchy marrow involvement, in which case, a repeat bone marrow biopsy may be indicated. The monotypic nature of the bone marrow plasma cells is established by the demonstration of an abnormal κ:λ ratio by immunohistochemistry or flow cytometry. Myeloma cells typically stain positive for CD38, CD56, and CD138, and are usually negative for surface immunoglobulin and CD19 on flow cytometry. Up to 20 % stain positively for CD20. Given the impact of multiple myeloma cell cytogenetic abnormalities on prognosis (see section “Risk Stratification”), bone marrow plasma cells should be studied using conventional karyotyping to detect hypodiploidy or deletion 13, and by FISH studies to detect specific chromosome abnormalities, such as t(11;14), t(4;14), t(14;16), t(6;14), t(14;20), hyperdiploidy, and deletion 17p [29–31]. In addition, if possible, gene expression profiling studies should be done to determine whether patients have high-risk features (see section “Risk Stratification”) [32].
Identification of Bone Disease
Plain radiographic examination of all bones, including long bones, (skeletal survey) is required for detecting lytic bone lesions in multiple myeloma. Conventional roentgenograms show skeletal abnormalities in almost 80 % of the patients with multiple myeloma; often these lesions have a characteristic punched-out appearance (Fig. 33.2). Osteoporosis and/or fractures may also be detected by conventional radiography. Rarely, osteosclerotic lesions can occur. CT and MRI scans are more sensitive than conventional radiography in detecting bone disease and are being increasingly used to detect lytic bone lesions in patients suspected of having multiple myeloma [33]. CT and/or MRI studies (Fig. 33.3) are also indicated when symptomatic areas show no abnormality on routine radiographs. Fluoro-deoxyglucose positron emission tomography (FDG-PET) combined with CT (PET-CT) is useful in the evaluation of bone disease and in the staging of multiple myeloma, particularly in patients without significant bone disease on conventional radiographs [34]. In contrast to the tests referred to earlier, the use of nuclear bone scans is not indicated, as this imaging modality is more sensitive for the detection of osteoblastic rather than osteolytic lesions. Bone mineral density studies are useful in identifying patients who do not have clear myeloma bone disease on radiographic studies, in order to identify patients at risk for pathologic fractures for whom prophylactic bisphosphonate therapy might be considered.
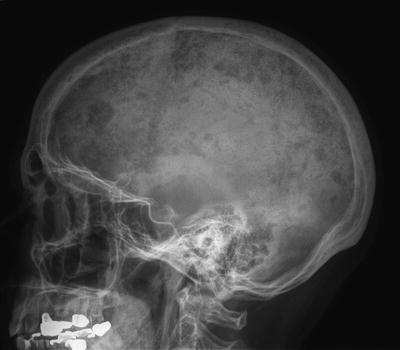
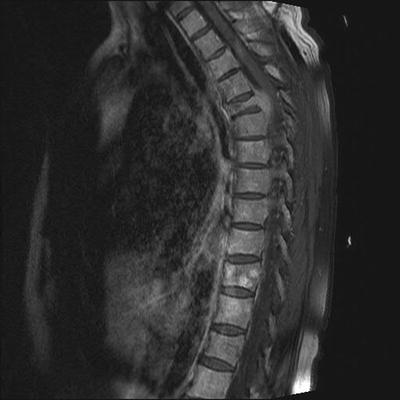
Fig. 33.2
Osteolytic lesions in the skull on plain radiographs in a patient with myeloma
Fig. 33.3
Magnetic resonance imaging (MRI) of a myeloma patient showing destruction of T5 vertebral body by a plasmacytoma
Laboratory Testing for Follow-Up and Monitoring
Once myeloma is diagnosed, patients require periodic measurements of M protein by SPEP and UPEP to assess treatment response, and monitor for relapse. Most patients without measurable disease in serum or urine, defined as less than 1 g/dL M protein on SPEP and <200 mg/24 h M protein on UPEP, can be followed using the serum FLC assay provided the FLC ratio is abnormal and the level of the involved FLC is ≥10 mg/dL [35]. Patients should also be monitored by periodic CBC, serum creatinine, and calcium measurements. In general, these laboratory tests and M protein measurements are done monthly during active therapy, and once every 3–4 months when the patients are being observed without therapy or using maintenance therapy. Radiographic tests are typically done every 6–12 months depending on response to treatment, as well as when symptoms indicate their need. Bone marrow studies are repeated if needed to confirm complete response or when clinically indicated to assess relapse. Response to therapy is done using the International Myeloma Working Group uniform response criteria (Table 33.2) [36].
Table 33.2
International Myeloma Working Group uniform response criteria for multiple myeloma
Response subcategory | Response criteria |
|---|---|
Complete responsea (CR) | •Negative immunofixation of serum and urine, and |
•Disappearance of any soft tissue plasmacytomas, and | |
•<5 % plasma cells in bone marrow | |
Stringent complete response (sCR)b | CR as defined previously plus |
•Normal FLC ratio, and | |
•Absence of clonal PC by immunohistochemistry or 2–4 color flow cytometry | |
Very good partial response (VGPR)a | •Serum and urine M-component detectable by immunofixation but not on electrophoresis, or |
•³90 % or greater reduction in serum M-component plus urine M-component <100 mg/24 h | |
Partial response (PR) | •³50 % reduction of serum M-protein and reduction in 24-h urinary M-protein by ≥90 % or to <200 mg/24 h |
•If the serum and urine M-protein are unmeasurable a ≥50 % decrease in the difference between involved and uninvolved FLC levels is required in place of the M-protein criteria | |
•If serum and urine M-protein are unmeasurable, and serum free light assay is also unmeasurable, ≥50 % reduction in bone marrow plasma cells is required in place of M-protein, provided baseline percentage was ≥30 % | |
•In addition to the previous criteria, if present at baseline, ≥50 % reduction in the size of soft tissue plasmacytomas is also required | |
Stable disease (SD) | •Not meeting criteria for CR, VGPR, PR or progressive disease |
Progressive disease (PD)b | •Increase of 25 % from lowest response value in any one or more of the following: |
Serum M-component (absolute increase must be ≥0.5 g/dL)c and/or | |
Urine M-component (absolute increase must be ≥200 mg/24 h) and/or | |
Only in patients without measurable serum and urine M-protein levels: the difference between involved and uninvolved FLC levels (absolute increase must be >10 mg/L) | |
Only in patients without measurable serum and urine M protein levels and without measurable disease by FLC levels, bone marrow plasma cell percentage (absolute % must be ≥10 %) | |
•Definite development of new bone lesions or soft tissue plasmacytomas or definite increase in the size of existing bone lesions or soft tissue plasmacytomas | |
•Development of hypercalcemia (corrected serum calcium >11.5 mg/dL) that can be attributed solely to the plasma cell proliferative disorder |
Differential Diagnosis
MGUS and SMM are differentiated from multiple myeloma and related disorders based on the presence or absence of end-organ damage that can be attributed to the plasma cell disorder (Table 33.1) [19]. Myeloma should also be differentiated from related plasma cell disorders such as solitary plasmacytoma, Waldenstrom’s macroglobulinemia, and light chain (AL) amyloidosis. Patients with MGUS may have renal dysfunction due to unrelated disorders or bone lesions due to an unrelated metastatic carcinoma and can be mistakenly diagnosed as myeloma. To be considered myeloma, the observed end-organ damage (anemia, hypercalcemia, renal failure, or bone lesions) must be felt to be attributable to the underlying plasma cell disorder. If there is any doubt, a renal biopsy or biopsy of suspected bone lesions is needed.
Risk Stratification
Survival in myeloma partially depends on disease stage, including the Durie–Salmon Stage (DSS) [37] or the International Staging System (ISS) [38, 39]. Although staging provided prognostic information for counseling, it is not useful for making therapeutic choices. The choice of therapy in myeloma is dependent on a risk stratification model that relies on a number of independent prognostic markers, particularly the molecular cytogenetic classification of the disease [40]. At the Mayo Clinic, newly diagnosed myeloma is stratified into standard-risk and high-risk disease using the Mayo stratification for myeloma and risk-adapted therapy (mSMART) (Fig. 33.4) [29, 31]. This stratification often guides treatment and refines the estimates for survival, with patients with standard risk having a median OS of 6–7 years, and those with high risk disease having a median OS of <2–3 years, even in patients having undergone previous tandem autologous stem cell transplantation (ASCT) [2].

Fig. 33.4
Risk stratification of myeloma
Treatment of Newly Diagnosed Myeloma
In the last decade, the treatment of myeloma has undergone remarkable advances with the emergence of thalidomide [41], bortezomib [42, 43], and lenalidomide [44, 45] as effective agents [46]. Bortezomib is a first-in-class proteasome inhibitor, and although the specific mechanisms of sensitivity and resistance are still unresolved, it is clear that the antimyeloma effect is related to the inhibition of the proteasome pathway [47–49]. The mechanism of action of thalidomide and lenalidomide is unclear, but both are felt to have immunomodulatory properties [50]. With the arrival of these new agents, the overall survival in myeloma has improved significantly in the last decade [51, 52].
Initial therapy depends on eligibility for ASCT and risk stratification [53]. Eligibility for stem cell transplantation is determined by age, performance status, and coexisting comorbidities. Risk stratification is based on the presence or absence of high-risk factors [29].
Myeloma is not considered by most investigators to be a curable malignancy, although some believe that with aggressive therapy a subset of patients can be maintained in a continuous remission. There is ongoing debate between whether the disease should be treated with an intention to cure versus a disease control strategy that employs active agents in a sequential manner [54]. An approach to the treatment of symptomatic newly diagnosed multiple myeloma is outlined in Fig. 33.5a, b. The overall approach to therapy of newly diagnosed myeloma based on transplant eligibility is discussed first. The specific strategies used for high-risk myeloma are discussed next. Table 33.3 lists the most common regimens used in the treatment of newly diagnosed myeloma. Patients with smoldering, asymptomatic myeloma should not receive therapy [70], but are candidates for clinical trials investigating experimental approaches such as lenalidomide or other agents with low toxicity [71, 72].
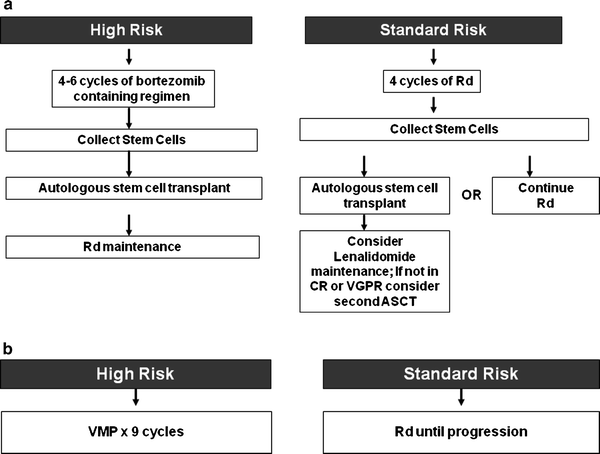
Fig. 33.5
(a) Approach to the treatment of newly diagnosed myeloma in transplant eligible patients. (b) Approach to the treatment of newly diagnosed myeloma in transplant ineligible patients. MPT melphalan–prednisone–thalidomide; VMP bortezomib, melphalan, prednisone; Rd lenalidomide plus low dose dexamethasone; CR complete response; VGPR very good partial response
Table 33.3
Major regimens for the treatment of newly diagnosed multiple myeloma
Regimen | Usual dosing schedulea |
|---|---|
Melphalan–Prednisone (MP) (7-day schedule) [1] | Melphalan 8–10 mg oral days 1–7 |
Prednisone 60 m/day oral days 1–7 | |
Repeated every 6 weeks until plateau as primary therapy | |
Melphalan–Prednisone (MP) (4-day schedule) [55] | Melphalan 0.25 mg/kg (9 mg/m [2]) oral days 1–4 |
Prednisone 2 mg/kg oral days 1–4 | |
Repeated every 4–6 weeks until plateau as primary therapy | |
Thalidomide 200 mg oral days 1–28 | |
Dexamethasone 40 mg oral days 1, 8, 15, 22 | |
Repeated every 4 weeks × 4 cycles as pre-transplant induction therapy; or continued till plateau or progression if used as primary therapy | |
Lenalidomide–Dexamethasone (Rev/low-dose Dex) [58] | Lenalidomide 25 mg oral days 1–21 every 28 days |
Dexamethasone 40 mg oral days 1, 8, 15, 22 every 28 days | |
Repeated every 4 weeks × 4 cycles as pre-transplant induction therapy; or continued till plateau or progression if used as primary therapy | |
Bortezomib-Dexb (Vel/Dex) [59] | Bortezomib 1.3 mg/m [2] intravenous days 1, 8, 15, 22 |
Dexamethasone 20 mg on day of and day after bortezomib (or 40 mg days 1, 8, 15, 22) | |
Repeated every 4 weeks × 4 cycles as pre-transplant induction therapy | |
Melphalan 0.25 mg/kg oral days 1–4 (use 0.20 mg/kg/day oral days 1–4 in patients over the age of 75) | |
Prednisone 2 mg/kg oral days 1–4 | |
Thalidomide 100–200 mg oral days 1–28 (use 100 mg dose in patients >75) | |
Repeated every 6 weeks × 12 cycles as primary therapy | |
Bortezomib 1.3 mg/m [2] intravenous days 1, 8, 15, 22 | |
Melphalan 9 mg/m [2] oral days 1–4 | |
Prednisone 60 mg/m [2] oral days 1–4 | |
Repeated every 35 days × 49 cycles as primary therapy | |
Bortezomib 1.3 mg/m [2] intravenous days 1, 8, 15, 22 | |
Thalidomide 100–200 mg oral days 1–21 | |
Dexamethasone 20 mg on day of and day after bortezomib (or 40 mg days 1, 8, 15, 22) | |
Repeated every 4 weeks × 44 cycles as pre-transplant induction therapy | |
Cyclophosphamide 300 mg/m [2] orally on days 1, 8, 15, 22 | |
Bortezomib 1.3 mg/m [2] intravenously on days 1, 8, 15, 22 | |
Dexamethasone 40 mg orally on days on days 1, 8, 15, 22 | |
Repeated every 4 weeks × 44 cycles as pre-transplant induction therapy cycles | |
Bortezomib 1.3 mg/m [2] intravenous days 1, 8, 15 | |
Lenalidomide 25 mg oral days 1–14 | |
Dexamethasone 20 mg on day of and day after bortezomib (or 40 mg days 1, 8, 15, 22) | |
Repeated every 3 weeks × 4 cycles as pre-transplant induction therapy |
Initial Therapy in Patients Eligible for ASCT
Prolonged melphalan-based therapy must be avoided in patients with newly diagnosed multiple myeloma who are considered eligible for ASCT since it can interfere with adequate stem cell mobilization, regardless of whether an early or delayed transplant is contemplated. Typically, patients are treated with approximately 2–4 cycles of induction therapy prior to stem cell harvest [73]. This includes patients who are transplant candidates, but who wish to reserve ASCT as a delayed option for relapsed refractory disease. Such patients can resume induction therapy following stem cell collection until a plateau phase is reached, reserving [65] ASCT for relapse. Thalidomide plus dexamethasone (TD) is approved for newly diagnosed myeloma in the United States, but currently the main options for initial therapy based on randomized controlled studies are lenalidomide plus low-dose dexamethaosone (Rd) [58, 74], bortezomib plus dexamethasone (VD) [75, 76], and bortezomib, thalidomide, dexamethasone (VTD) (Table 33.4). In addition, bortezomib, lenalidomide, dexamethasone (VRd) [67, 69] and cyclophosphamide, bortezomib, dexamethasone (CyBorD) [68] are increasingly considered as other options and are also discussed.
Table 33.4
Results of recent randomized studies in newly diagnosed myeloma patients eligible for transplantation
Trial | Regimen | No. of patients | Overall response rate (%) | CR plus VGPR (%) | Progression-free survival (median in years) | P value for progression-free survival | 3 year overall survival rate (%)a | Overall survival (median in years) | P value for overall survival |
|---|---|---|---|---|---|---|---|---|---|
Rajkumar et al. [58] | RD | 223 | 81 | 50 | 19.1 | 0.026 | 75 | NR | 0.47 |
Rd | 222 | 70 | 40 | 25.3 | 74 | NR | |||
VAD | 218 | 65 | 16 | 30 | 0.057 | 80 | NR | 0.46 | |
VD | 223 | 82 | 39 | 36 | 80 | NR | |||
TD | 238 | 80 | 31 | NR | 0.009 | N/A | N/A | ||
VTD | 236 | 93 | 62 | NR | N/A | N/A | |||
Moreau et al. [76] | VD | 99 | 81 | 35 | N/A | N/A | N/A | ||
VTD | 100 | 90 | 51 | N/A | N/A | N/A |
Thalidomide–Dexamethasone
The up-front use of Thal/Dex was initially based on three phase II clinical trials [79–81]. Response rates with Thal/Dex ranged from 64 to 76 % in these studies. The Eastern Cooperative Oncology Group (ECOG) reported the results of a randomized trial comparing Thal/Dex to dexamethasone in 202 patients [56]. The best response within four cycles of therapy was significantly higher with Thal/Dex compared to dexamethasone alone; 63 % versus 41 %, respectively, P = 0.0017. Stem cell harvest was successful in 90 % of the patients in each arm. Deep vein thrombosis (DVT) was more frequent with Thal/Dex (17 % versus 3 %). Overall, grade 3 or higher non-hematologic toxicities were seen in 67 % of the patients within four cycles with Thal/Dex, and 43 % with dexamethasone alone (P < 0.001). Early mortality (first 4 months) was 7 % with Thal/Dex and 11 % with dexamethasone alone. Based on this trial, the United States Food and Drug Administration (FDA) granted accelerated approval for Thal/Dex for the treatment of newly diagnosed myeloma. Another randomized, double-blind, placebo-controlled study compared Thal/Dex versus dexamethasone alone as primary therapy in 470 patients with newly diagnosed myeloma [57]. As in the ECOG trial, response rates were significantly higher with Thal/Dex compared to placebo/dex, 63 % versus 46 %, respectively, P < 0.001. The TTP also was superior with Thal/Dex versus placebo/dex, 22.6 versus 6.5 months, P < 0.001. DVT and other grade 3–4 events were more frequent with Thal/Dex (30.3 % versus 22.8 %). In a recent Mayo Clinic study of 411 newly diagnosed patients including a case-matched analysis, Gay and colleagues found that lenalidomide plus dexamethasone was significantly superior to thalidomide–dexamethasone in terms of response rate, progression-free survival, and overall survival [82]. TD remains an option when lenalidomide is not available for initial therapy, with or without the addition of cyclophosphamide. Patients receiving thalidomide-based regimens require DVT prophylaxis with aspirin, low-molecular weight heparin, or coumadin [83, 84].
Lenalidomide-Low Dose Dexamethasone (Rd)
Lenalidomide is a thalidomide analogue that is safer and more effective than thalidomide. It is currently approved by the FDA for the treatment of multiple myeloma in patients who have failed one prior therapy. In newly diagnosed multiple myeloma, a phase II trial conducted at the Mayo Clinic demonstrated remarkably high activity with lenalidomide plus high dose dexamethasone. Thirty-one of 34 patients (91 %) achieved an objective response [44]. With longer follow up, 56 % of the patients achieved VGPR or better [85]. The rates of grade 3 or higher non-hematologic toxicity (50 %) are lower than with thalidomide plus dexamethasone. The OS survival rate at 3 years was remarkably high at 88 % [85].
Preliminary results from a randomized trial showed that lenalidomide plus dexamethasone was superior to dexamethasone alone in newly diagnosed myeloma [74]. A recent randomized trial (E4A03) compared lenalidomide plus high dose dexamethasone (RD) which used dexamethasone at 40 mg days 1–4, 9–12, 17–20) versus lenalidomide plus low dose dexamethasone (Rd) which used dexamethasone 40 mg once weekly) [58]. The study showed lower toxicity, lower early mortality, and superior overall survival with Rd compared with RD, with 1 year overall survival rate of 96 % versus 87 %, respectively (P < 0.001). Deep vein thrombosis (DVT) rates were also lower with Rd making this one of the safest pre-transplant induction regimens for multiple myeloma. As a result, the trial was closed early at a median follow up of 12 months, and all patients crossed over to Rd. Overall results with median follow up of 36 months are shown in Table 33.4. Following this study, the use of high-dose dexamethasone regimens in newly diagnosed myeloma is no longer recommended. All recent newly diagnosed trials have used low -dose dexamethasone approaches, particularly when using novel agent combinations
Rd induces rapid responses, is well tolerated, and does not carry the risk of neuropathy making it an excellent choice for initial therapy. However, Rd may impair collection of peripheral blood stem cells for transplant in some patients when mobilized with granulocyte stimulating factor (G-CSF) alone [86, 87]. Stem cell mobilization in these patients is usually successful with a chemotherapy containing mobilization regimen such as cyclophosphamide and G-CSF. Plerixafor, a CXCR4 (a chemokine receptor) inhibitor, is a stem cell mobilizing agent that usually allows adequate stem cell collection in the subset of patients who have difficulty mobilizing with G-CSF alone. We typically offer initial collection with chemotherapy or plerixafor to elderly patients over the age of 65 and to patients who receive more than four cycles of Rd therapy [73, 88]. Rd also carries the risk of DVT and all patients require antithrombosis prophylaxis with aspirin, low-molecular weight heparin, or coumadin [83, 84].
Bortezomib–Dexamethasone
Bortezomib is a proteasome inhibitor approved for the treatment of patients with relapsed and refractory multiple myeloma. In newly diagnosed multiple myeloma, bortezomib alone results in responses in approximately 40 % [89]. Bortezomib has also shown single-agent activity in a clinical trial conducted in patients with high-risk newly diagnosed myeloma [90]. Significantly higher response rates (approximately 70–90 %) have been observed with bortezomib plus dexamethasone (VD) [59, 91]. No adverse effect on stem cell mobilization has been reported with VD. The most important grade 2 or higher adverse events are sensory neuropathy (31 %) and fatigue (25 %) [91]. Harousseau and colleagues reported preliminary results of a randomized trial conducted by the Intergroupe Francophone du Myélome (IFM) comparing vincristine, Adriamycin, dexamethasone (VAD) versus VD as pre-transplant induction therapy [75, 77]. Patients were additionally randomized for consolidation therapy with or without two additional cycles of dexamethasone, cyclophosphamide, etoposide, and cisplatin (DCEP). All patients were then expected to proceed to ASCT. With over 400 patients enrolled, preliminary results show superior response rates and progression-free survival (PFS) with VD compared with VAD (Table 33.4).
The major drawback of VD is the need for intravenous therapy and the risk of neurotoxicity early in the disease course. The neuropathy with bortezomib can occur abruptly, and can be significantly debilitating in a subset of patients. Recent studies show that reducing the dose of bortezomib to once a week shows similar efficacy with significantly lower risk of neurotoxicity, with grade 3 or higher neuropathy rates decreasing from approximately 15 % to 2–3 % [62, 63]. As a result, once-weekly bortezomib is preferred in most patients for initial therapy, unless an urgent need for rapid disease control is felt [92]. Bortezomib-based regimens such as VD are of particular value in patients with renal failure [93] and in patients with high-risk multiple myeloma (see text to come). Unlike lenalidomide, bortezomib does not appear to have any adverse effect on stem cell mobilization [94].
Bortezomib–Thalidomide–Dexamethasone
Bortezomib, thalidomide, dexamethasone (VTD) has been tested against thalidomide–dexamethasone (TD), as well as against VD in separate randomized trials (Table 33.4). Cavo et al. compared pre-transplant induction therapy with VTD to TD [65, 78]. VTD was associated with better response rates and PFS. Post ASCT, the CR and near CR rates were significantly higher with VTD. However, VTD was associated with a higher incidence of skin rash and neuropathy. In a recent randomized trial by the IFM, Moreau et al. showed that reduced bortezomib-dose VTD (termed vTD by the authors) resulted in better pre-transplant response rates compared with VD [76].
Although the results described previously are preliminary, they do suggest that three-drug combinations can improve response rates and PFS compared with two-drug combinations. However, there are no data on whether the early incorporation of the third drug results in prolongation of overall survival, and what effect adding the third drug has on quality of life, especially given the neuroxicity associated with bortezomib and thalidomide.
Bortezomib–Lenalidomide–Dexamethasone
Bortezomib, lenalidomide, dexamethasone (VRD) produces high overall and complete response rates when used in the treatment of newly diagnosed multiple myeloma. In one phase II trial, 100 % of the patients achieved partial response, and 67 % achieved very good partial response (VGPR) or better [69]. The most common toxicities included sensory neuropathy (80 %) and fatigue (64 %), including 29 % with grade 2 or higher neuropathy. Similar results have been reported from another phase II trial [67]. A Southwest Oncology Group (SWOG) randomized trial is currently comparing VRd to Rd in the United States.
Cyclophosphamide–Bortezomib–Dexamethasone
A three-drug combination of cyclophosphamide, bortezomib, and dexamethasone (CyBorD) has shown significant activity in newly diagnosed multiple myeloma [68]. In a study of patients, the overall response rate was 88 %, with 61 % of VGPR or better, and 39 % complete/near complete response (CR/nCR). As with other bortezomib-based regimens, the main adverse event of concern is peripheral neuropathy, and weekly administration of bortezomib is favored [92]. Preliminary results from a recent randomized trial showed similar or better activity compared with VRd making this a promising regimen for further testing.
VDT-PACE
Another approach used in the total therapy protocols from Arkansas is to use multi-agent combination chemotherapy, such as VDT-PACE (bortezomib, dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, and etoposide) [95, 96]. In the absence of randomized trials, it is not clear whether VDT-PACE is superior to other less aggressive combinations such as VRd or CyBorD, especially since the added value of cisplatin, etoposide, and even doxorubicin in myeloma is unclear. However, VDT-PACE may be an important option for patients with very aggressive disease such as plasma cell leukemia or multiple extramedullary plasmacytomas.
Other Regimens
Several other regimens have been tested in newly diagnosed multiple myeloma, but there are no clear data from randomized controlled trials that they have an effect on long-term endpoints compared with the regimens discussed earlier. One strategy that has been explored is to add doxorubicin or liposomal doxorubicin to either thalidomide or lenalidomide. Another strategy that is promising is to combine all effective agents in an attempt to achieve the highest CR rate upfront. The Evolution trial has shown that the combination of bortezomib, dexamethasone, cyclophosphamide, lenalidomide, and VDCR produces responses in nearly all patients with newly diagnosed myeloma, including 68 % VGPR or better [97]. The randomized phase II portion of this study is comparing the efficacy of VDCR, VRD, and CyBorD.
Older Regimens
VAD, used for many years as pre-transplant induction therapy, is no longer recommended [98]. Cavo and colleagues, in a matched case–control study of 200 patients, demonstrated that response rates with VAD were significantly lower compared to Thal/Dex; 76 % versus 52 %, respectively [99]. Preliminary results from a randomized trial from France as discussed earlier confirm these findings [100]. High-dose pulsed dexamethasone alone dosed at 40 mg orally, on days 1–4, 9–12, and 17–20 every 4–5 weeks has been used in the past as initial therapy, but is also no longer recommended. Objective response rates with high-dose dexamethasone are low, approximately 45 % [56]. More importantly, in randomized trials, the early mortality rate associated with dexamethasone is over 10 % within the first 4 months of therapy, reflecting the high toxicity of this regimen [57, 101].
Initial Therapy in Patients Not Eligible for ASCT
In patients with newly diagnosed multiple myeloma who are considered ineligible for ASCT due to advanced age, poor performance status, heart failure, liver failure, or other comorbidities, the major options at present are either melphalan-based combination therapies or Rd (Table 33.5) [106]. With melphalan-based therapy, patients are usually treated for a fixed duration of time (9–18 months) and then observed. With Rd, it is unclear whether treatment should continue until relapse or be stopped after a fixed duration of therapy.
Table 33.5
Results of recent randomized studies in newly diagnosed myeloma patients not eligible for transplantation
Trial | Regimen | No. of patients | Overall response rate (%) | CR plus VGPR (%) | Progression-free survival (median in years) | P value for progression-free survival | 3 year overall survival rate (%)a | Overall survival (median in years) | P value for overall survival |
|---|---|---|---|---|---|---|---|---|---|
Facon et al. [60] | MP | 196 | 35 | 7 | 17.8 | <0.001 | 48 | 33.2 | <0.001 |
Mel 100 | 126 | 65 | 43 | 19.4 | 52 | 38.3 | |||
MPT | 125 | 76 | 47 | 27.5 | 66 | 51.6 | |||
Hulin et al. [61] | MP + Placebo | 116 | 31 | 7 | 18.5 | 0.001 | 40 | 29.1 | 0.028 |
MPT | 113 | 62 | 21 | 24.1 | 55 | 44 | |||
Wijermans et al. [102] | MP | 168 | 45 | 10 | 9 | <0.001 | 43 | 31 | 0.05 |
MPT | 165 | 66 | 27 | 13 | 55 | 40 | |||
Palumbo et al. [103] | MP | 164 | 48 | 11 | 14.5 | 0.004 | 65 | 47.6 | 0.79 |
MPT | 167 | 69 | 29 | 21.8 | 65 | 45 | |||
Waage et al. [104] | MP + Placebo | 175 | 33 | 7 | 14 | NS | 43 | 32 | 0.16 |
MPT | 182 | 34 | 23 | 15 | 43 | 29 | |||
MP | 331 | 35 | 8 | 16.6 | <0.001 | 54 | 43 | <0.001 | |
VMP | 337 | 71 | 41 | 24 | 69 | NR |
Melphalan, Prednisone, Thalidomide
Four randomized studies have shown that melphalan, prednisone, thalidomide (MPT) improves response rates compared to MP [60, 61, 102, 104, 107]. Four of these trials have shown a significant prolongation of PFS with MPT [60, 61, 102, 107], and an OS advantage has been observed in the two IFM trials and in the trial by Wijermans et al. (Table 33.5) [60, 61, 102, 104, 107].
Palumbo et al. from the Italian Multiple Myeloma Network Gruppo Italiano Malattie EMatologiche dell’Adulto (GIMEMA) randomized MP for 6 months versus MPT for 6 months followed by maintenance thalidomide [107]. Overall response rates were significantly higher with the MPT compared to MP (76 % versus 48 %) as was the CR plus near CR rate (28 % versus 7 %). MPT resulted in superior 2-year progression-free survival (PFS) rates (54 % versus 27 %, P < 0.001), and a trend toward an improved 3-year overall survival. The first IFM trial led by Facon et al. randomized 447 patients (ages 65–75) to MP versus MPT versus tandem ASCT with reduced dose melphalan at 100 mg/m [2] (Mel 100) [60]. PFS was superior with MPT compared to either MP or tandem Mel 100 groups, P < 0.001. There was also a significant survival advantage with MPT, with median overall survival of 52 months, 33 months, and 38 months, respectively. Early (first 3 months) mortality rate was 8 % with MP compared to 3 % with MPT. In the second IFM trial, Hulin et al. confirmed a survival advantage with MPT compared to MP in a randomized trial in 229 patients over the age of 75, with median survival of 44 versus 29 months, respectively, P = 0.028 [61]. This trial showed that the benefit of MPT over MP persists in patients up to at least age 85, but it is important to note that patients received a lower dose of melphalan and thalidomide (Table 33.3). A recent report by Wijermans et al. on behalf of the Dutch-Belgium Hemato-Oncology Cooperative Group (HOVON) confirms the superiority of MPT over MP [102]. In contrast to these four trials, Waage and colleagues from the Nordic Myeloma Study Group found no PFS or overall survival benefit with MPT compared with MP in a randomized placebo controlled trial [104]. The reasons for this discrepancy are not clear, but may be related to the use of high doses of thalidomide (400 mg) in an elderly patient population resulting in increased early mortality particularly among those over the age of 75. A recent meta-analysis of these five studies shows a clear superiority of MPT in terms of PFS, and a trend towards improved survival [108].
As a result of the previous studies, MPT is an option for patients not eligible for ASCT. However, MPT is associated with greater toxicity than MP or Rd, and not all elderly patients may be able to tolerate the regimen. Grade 3–4 adverse events occur in approximately 55 % of the patients treated with MPT, compared to 22 % with MP [107]. As with Thal/Dex, there is a significant (20 %) risk of DVT with MPT in the absence of thromboprophylaxis. However, this rate drops to approximately 3 % with the use of thromboprophylaxis (e.g., enoxaparin) [107].
Bortezomib, Melphalan, Prednisone
Mateos et al. found a response rate of 89 %, with a remarkably high CR rate of 32 % with the Bortezomib, Melphalan, Prednisone (VMP) regimen in a phase II study [109]. VMP was compared with MP in a large phase III trial of 682 patients, median age 71 years [64]. VMP had a significantly superior response rate compared to MP (71 % versus 35 %, P < 0.001), as well as a superior CR rate (30 % versus 4 %). TTP was significantly superior with VMP compared to MP, 24 versus 17 months, respectively, P < 0.001. Importantly, VMP appeared to overcome adverse cytogenetic features; there was no significant difference in TTP or overall survival with VMP in patients with and without high-risk myeloma. Updated results from this trial have been published and are shown in Table 33.5 [105]. Neuropathy is a significant risk with VMP therapy; grade 3 neuropathy occurred in 13 % of the patients versus 0 % with MP [64]. Neuropathy of any grade with VMP occurred in 44 % of the patients; grade 2 or higher in 30 %. As discussed earlier, the risk of neuropathy can be greatly decreased by a once-weekly schedule of bortezomib [62, 63]. Consequently, once-weekly bortezomib is preferred in most patients for initial therapy, unless an urgent need for rapid disease control is felt. VMP is an alternative to MPT for the treatment of patients with newly diagnosed myeloma not eligible for ASCT, especially those with high-risk features (Fig. 33.5b).
Lenalidomide–Low Dose Dexamethasone (Rd)
Although developed initially as a pre-transplant induction regimen, this has quickly become one of the most commonly used regimens for the treatment of elderly patients with newly diagnosed myeloma because of its excellent tolerability, convenience, and efficacy (Fig. 33.5b). In the E4A03 randomized trial of RD versus Rd, the benefit of lowering dexamethasone dose was most apparent in patients over the age of 65 [58]. Jacobus et al. [110] recently provided preliminary data on the safety and efficacy of Rd in elderly patients aged 70 years and above treated on the ECOG E4A03 trial. Among 147 patients aged 70 years and above the median PFS was 22 months with Rd versus 16 months with RD, P = 0.11. Overall survival was significantly superior with Rd compared with RD, P = 0.03; 3-year overall survival 73 % versus 61 %, respectively. The 3-year overall survival rate with Rd in this subset excluding patients who underwent ASCT was 70 %, comparable to those seen with MPT and VMP. Gay et al. found similar results in a cohort of 42 newly diagnosed elderly myeloma patients aged 70 years and above [111], with a 3-year overall survival rate of 70 %. Randomized phase III studies, comparing Rd with alkylator-based combinations such as MPT or MPV, are needed. One ongoing trial is currently comparing MPT versus Rd for 18 months versus Rd until progression.
Other Regimens
Melphalan plus prednisone (MP) has been used in the treatment of multiple myeloma for over 40 years, and may still have a role in elderly patients who do not have access to Rd in whom therapy with MPT or VMP is not considered safe or feasible [112, 113]. The response rate with MP is approximately 50 %. The median survival is approximately 3–4 years. A meta-analysis of 26 randomized trials found superior response rates but no survival benefit with combination chemotherapy regimens compared with MP prior to the arrival of thalidomide and bortezomib [113]. MP is well tolerated, but responses can sometimes take 6 months to occur. The substitution of dexamethasone in place of prednisone improves response rate and the speed of response, but does not improve the overall survival.
Palumbo and colleagues have studied the addition of lenalidomide to MP (MPR) in newly diagnosed patients >65 years of age [114]. Fifty-four patients were studied. The overall response rate was 85 %, with 42 % of the patients achieving at least VGPR or better, and 17 % of the patients achieving a CR. Major grade 3–4 adverse events were neutropenia (66 %), thrombocytopenia (34 %), anemia (17 %), rash (10 %), and febrile neutropenia (8 %). Results of a recent phase III trial show improved PFS with MPR followed by lenalidomide maintenance (MPR-R) compared with MP or MPR without maintenance [115]. However, it is not clear why a PFS advantage was not seen with MPR versus MP in the absence of lenalidomide maintenance. An ECOG randomized trial (E1A06) is currently comparing MPR to MPT.
Hematopoietic Stem Cell Transplantation
Autologous Stem Cell Transplantation
Although not curative, ASCT improves CR rates and prolongs median OS in multiple myeloma by approximately 12 months [116–119]. The mortality rate with ASCT is very low at 1–2 %, and a substantial proportion of transplants can be done on an outpatient basis [120]. Melphalan at 200 mg/m [2] (Mel 200) is the most widely used preparative (conditioning) regimen for ASCT. Melphalan 140 mg/m [2] and 8 Gy total body irradiation (TBI) is inferior and is not recommended [121]. In a recent phase 3 study, 298 patients were randomized to tandem ASCT with Mel 200 or Mel 100 [122]. The CR rate was higher with Mel 200 compared with Mel 100, 15 % versus 8 %. The median PFS was also superior, 31.4 versus 26.2 months, respectively, P = 0.01. Mel 200 did not carry an increase in treatment-related mortality, and although, the overall survival was similar between the two arms, Mel 200 is still considered the standard induction for myeloma. Mel 100 can be considered in elderly patients and in patients who have renal dysfunction. Studies are ongoing to determine if the conditioning regimen can be improved with the addition of radioactive compounds (Holmium (HO [123] DOTMP), Samarium 153-SM-EDTMP) [124, 125], or bortezomib.
Three randomized trials show that survival is similar whether ASCT is done early (immediately following four cycles of induction therapy) or delayed (at the time of relapse as salvage therapy) [126–128]. In the Spanish Programa Español Tratamientos en Hematología (PETHEMA) group randomized trial, patients responding to induction therapy had similar OS and PFS when ASCT was compared with eight cycles of chemotherapy [129]. The results of this trial suggest that the greatest benefit from ASCT may be in those with disease refractory to induction therapy [130, 131].
There is little doubt that ASCT prolongs survival in multiple myeloma, but its timing (early versus delayed) is controversial [53, 118]. There are three randomized trials that show that early versus delayed transplantations done at first relapse are identical in terms of overall survival [126–128]. Although these trials were done prior to the arrival of novel agents, there are no data that overall survival is superior with early transplant. Given the various new, effective and well-tolerated agents to treat multiple myeloma, some patients and physicians often choose to delay the procedure. The timing is dependent on response and toxicity to initial therapy, age of patient, and patient and physician preference. An ongoing randomized trial is comparing VRD induction followed by ASCT versus continued VRD therapy.
Tandem Autologous Transplantation
With tandem (double) ASCT, patients receive a second planned ASCT after recovery from the first procedure [132, 133]. The IFM 94 randomized trial found significantly better PFS and overall survival in recipients of double versus single ASCT [134]. A similar benefit was also demonstrated in a randomized trial conducted in Italy [135]. However, other randomized trials have failed to show significant improvement in overall survival with tandem ASCT, but they have shorter follow up [136, 137]. A recent meta-analysis did not find a substantial benefit in overall survival with tandem ASCT [138]. In both the French and Italian trials, the benefit of a second ASCT was primarily in patients failing to achieve a CR or VGPR with the first procedure. Based on these results, we recommend that stem cells adequate for two transplants be collected early in the disease course in all eligible patients. Patients not achieving such a response are offered a second ASCT, while those in CR or VGPR can postpone consideration of the second ASCT until relapse [139]. A trial by the Bone Marrow Transplant Clinical Trials Network (BMT-CTN) group is currently testing tandem ASCT versus single ASCT versus single ASCT followed by VRD in a prospective randomized trial.
Allogeneic Transplantation
Studies have assessed the role of allogeneic transplantation in myeloma, prompted by the presence of a graft-versus-myeloma effect [140, 141]. However, only 5–10 % of the patients are candidates because of age, availability of a HLA matched sibling donor, and adequate organ function. The high treatment-related mortality (TRM), mainly related to graft versus host disease (GVHD), has made conventional allogeneic transplants unacceptable for most patients with multiple myeloma.
Several recent trials have been conducted using non-myeloablative allografting regimens (mini-allogeneic transplantation; reduced intensity conditioning allogeneic transplantation) [142]. Initial trials evaluating this approach included relapsed or refractory patients, which were thought to account at least in part for the poor outcomes. It also became apparent that this approach was less useful in patients who had significant residual tumor burden at the time of the non-myeloablative transplant, which led to the concept of a planned autologous SCT followed by a planned reduced intensity conditioning allogeneic stem cell transplant (RIC SCT) a couple of months later [143]. The initial studies had treatment-related mortalities approaching 25 %, and 3-OS and PFS rates were 41 % and 21 %, respectively [143]. Adverse OS was associated with chemoresistant disease, more than one prior transplantation, and absence of chronic GVHD.
Recent studies have used a planned tandem ASCT followed by non-myeloablative allograft approach, and have produced better outcomes with TRM of approximately 15 %, and 2-year OS of approximately 75 % [144, 145]. Among 102 patients (median follow up 6.3 years) treated with ASCT followed by a non-myeloablative allograft (2-Gy total body irradiation, with or without fludarabine) from a HLA identical sibling, the median TTP was 5 years, considerably longer than that reported with tandem ASCT [146]. Post allograft immunosuppression was with cyclosporine or tacrolimus plus mycophenolate mofetil. Still, 42 % developed grade 2–4 acute GVHD, 74 % developed extensive chronic GVHD, and the 5-year TRM was 18 %, almost all related to GVHD or infections. The 5-year overall survival rate was 64 % and 5-year PFS was 36 %. The dramatically lower 5-year PFS compared with the 5-year TTP rate illustrates the high TRM associated with this strategy since such a discrepancy is not seen with ASCT or conventional chemotherapy approaches in myeloma.
Garban et al. reported on one randomized trial of allogeneic stem cell transplantation in high-risk patients, defined by the presence of deletion 13 by FISH and beta-2 microglobulin > 3 mg/L [147]. Based on biological randomization, the patients were allocated to either double ASCT or to a single ASCT followed by a RIC allogeneic SCT. Although the patients did better than expected in both arms, with median OS of 41 and 35 months, respectively, there was no significant difference between the two arms with a median follow-up of 24 months. The major criticisms of this study have been that the criteria used to select ‘high-risk’ disease did not select for the highest risk patients and that the reduced intensity conditioning used may have been highly immunosuppressive, thereby abrogating the graft versus myeloma effect.
In contrast to the study referred to earlier, Bruno and colleagues found a significant overall survival advantage with a strategy of ASCT followed by non-myeloablative allograft [148]. Among 162 consecutive patients, those with an HLA-identical sibling received non-myeloablative allograft following ASCT. Patients without an HLA-identical sibling received tandem ASCT. On intent to treat basis, the median overall survival and PFS were longer in the 80 patients with HLA-identical siblings compared with the 82 patients without HLA-identical siblings (80 months versus 54 months, P = 0.01; and 35 months versus 29 months, P = 0.02, respectively). Results were similar when the study was analyzed based on actual treatment administered. As in other studies, the cumulative incidence rates of grades 2–4 GVHD was in excess of 40 %. Similar promising results have been reported by Gahrton et al. in a prospective trial comparing non-myeloablative allograft (n = 107) versus ASCT (n = 251), with arm assignment based on the availability of an HLA identical sibling donor [149]. Tandem ASCT was permitted in the ASCT arm and 122 patients received tandem ASCT. The conditioning regimen for the non-myeloablative allograft consisted of fludarabine 30 mg/m [2] × 3 plus TBI 2 Gy. On intent to treat basis, 2 year TRM was 13 % with allografting, and 5 % with ASCT, P = 0.014. The PFS rate at 5 years was 35 % (95 % CI 27–45 %) versus 18 % (95 % CI 14–24 %), respectively, P < 0.05. Overall survival was not significantly different, 65 % versus 57 %, respectively.
In contrast to the two trials discussed earlier, Rosinol and colleagues found no benefit in overall survival or PFS with an ASCT-allograft approach compared with tandem ASCT [150]. They treated 110 patients with multiple myeloma failing to achieve at least near-complete remission after a first ASCT with either non-myeloablative allografting (n = 25) or second ASCT (n = 85) depending on the availability of a HLA identical sibling donor. The TRM was 16 % versus 5 %, respectively, P = 0.07.
Given these conflicting results, the role of non-myeloablative-allogeneic transplantation is controversial, and remains investigational. The TRM (10–20 %) and GVHD rates with non-myeloablative allogeneic transplantation are unacceptably high [151]. Non-myeloablative allogeneic transplantation should only be considered in the context of clinical trials, especially in standard-risk multiple myeloma. Results of a BMT CTN trial that compared tandem ASCT versus ASCT followed by a non-myeloablative allogeneic transplantation are awaited.
Treatment Approach for Newly Diagnosed High-Risk Myeloma
Patients with high-risk multiple myeloma tend to do poorly with a median OS of approximately 2 years, even with tandem ASCT. Options for these patients should include novel therapeutic strategies [2, 29]. The treatment of high-risk patients varies from standard-risk patients in at least three ways: high-risk patients should consider early use of bortezomib, routine use of maintenance therapy, and target CR as a defined goal of therapy [31].
Bortezomib-containing regimens should be considered as initial therapy, especially for patients with t4;14 translocation, karyotypic deletion 13, or hypodiploidy. In several studies, bortezomib appears to overcome the adverse effect of these high-risk features [95, 109, 152, 153]. Thus for initial therapy in transplant candidates, a bortezomib-containing regimen such as VRd or CyBorD should be considered; similarly for non-transplant candidates, a bortezomib-based initial therapy such as VMP is preferred over Rd or MPT [31]. If ASCT is done (early or delayed), routine maintenance therapy should be considered given the high risk for early relapse. Finally, CR should be targeted as a therapeutic goal in high-risk myeloma. In contrast to standard-risk patients in whom survival is similar regardless of the achievement of CR, in high-risk patients, achievement of CR appears to be critical for long-term survival [154].
In addition to the approaches discussed previously, allogeneic transplantation may be an option in selected patients. However, as mentioned previously, the IFM 99 trial in patients with deletion 13 and high β2-microglobulin levels did not show significant benefit with this strategy compared to tandem ASCT [147]. Clearly, clinical trials and new agents specifically designed for high-risk multiple myeloma are needed, and enrolling patients in clinical trials investigating novel agents as initial therapy should be considered.
Maintenance Therapy
Maintenance therapy with interferon-α is of limited value and is seldom used [155]. Results from the Phase III US Intergroup Trial S9321 showed no benefit with interferon as maintenance therapy after ASCT [128]. A study by Berenson and colleagues suggested that prednisone may be useful for maintenance therapy [156]. Progression-free survival (14 versus 5 months) and OS (37 versus 26 months) were significantly longer with 50 mg versus 10 mg of prednisone orally every other day. As this comparison included only those who responded initially to steroid-based therapy, and patients did not receive ASCT, it is difficult to generalize these results to current practice.
Thalidomide
A French trial (IFM 99-02) randomized 597 patients (age <65) following tandem ASCT to no maintenance (arm A), pamidronate (arm B), or pamidronate plus thalidomide (arm C) [157]. There was a significant improvement in EFS with maintenance thalidomide plus pamidronate; the 3-year EFS rate from the time of randomization was 36 % in arm A, 37 % in arm B, and 52 % in arm C (P < 0.009). The corresponding 4-year OS rates were 77 %, 74 %, and 87 %, respectively (P < 0.04).
Spencer et al. randomized patients post ASCT to indefinite prednisolone maintenance therapy (n = 129) versus the same in addition to 12 months of thalidomide consolidation (n = 114). After a median follow-up of 3 years, the 3-year PFS was superior with thalidomide plus prednisolone compared with prednisolone alone, 42 % versus 23 %, respectively, P < 0.001. Overall survival at 3 years was also better, 86 % and 75 %, respectively, P = 0.004. Results of one other study also showed benefit with thalidomide maintenance, but the publication of this study has been retracted [158]. At present, thalidomide maintenance can be considered if access to lenalidomide is not available (see discussion to come) in patients who have failed to achieve VGPR or CR after transplant, and in high-risk patients
Lenalidomide
Two recent studies have shown prolongation of remission duration when lenalidomide was used post ASCT as maintenance therapy. Attal and colleagues from the IFM group treated patients (n = 614) less than 65 years of age with non-progressive disease after a first-line ASCT with lenalidomide consolidation (25 mg days 1–21 every 28 days) for 2 months followed by randomization to maintenance therapy with either lenalidomide (10–15 mg/day) or placebo (double-blind) [159]. PFS was significantly superior with lenalidomide maintenance, and this improvement occurred without any significant improvement in depth of response; median PFS 41 versus 23 months respectively, P < 0.001. This benefit was observed regardless of CR or VGPR status after ASCT. No significant differences in overall survival are apparent so far. In another study, the Cancer and Acute Leukemia Group B (CALGB) in the United States enrolled 568 patients who were treated first with ASCT (Mel 200 conditioning) followed by randomization to maintenance lenalidomide versus placebo (double-blind) [160]. The starting dose of lenalidomide was 10 mg/day, escalated to 15 mg/day after 3 months. The median PFS was significantly longer with lenalidomide maintenance compared with placebo, 46 versus 27 months, respectively, P < 0.001. An improvement in overall survival was noted, P = 0.03. In both studies, lenalidomide maintenance was associated with a two-fold higher risk of secondary malignancies. At this point until further data emerge lenalidomide maintenance is not considered standard of care, but these results need to be discussed with the patient along with the pros and cons of lenalidomide maintenance versus lenalidomide therapy at first relapse.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


