Acute myeloid leukemia with recurrent genetic abnormalities
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 (AML1-ETO) [M2]
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 [M4Eo]
APL with t(15;17)(q22;q12); PML-RARA [M3]
AML with t(9;11)(p22;q23); MLLT3-MLL
AML with t(6;9)(p23;q34); DEK-NUP214
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
Provisional entity: AML with mutated NPM1
Provisional entity: AML with mutated CEBPA
Acute myeloid leukemia with myelodysplasia-related changes
Therapy-related myeloid neoplasms
Acute myeloid leukemia, not otherwise specified
AML with minimal differentiation [M0]
AML without maturation [M1]
AML with maturation [M2]
Acute myelomonocytic leukemia [M4]
Acute monoblastic/monocytic leukemia [M5]
Acute erythroid leukemia [M6]
Pure erythroid leukemia
Erythroleukemia, erythroid/myeloid
Acute megakaryoblastic leukemia [M7]
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis
Myeloid sarcoma
Myeloid proliferations related to Down syndrome
Transient abnormal myelopoiesis
Myeloid leukemia associated with Down syndrome
Blastic plasmacytoid dendritic cell neoplasm
Acute leukemias of ambiguous lineage
Acute undifferentiated leukemia
Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1
Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged
Mixed phenotype acute leukemia, B-myeloid, NOS
Mixed phenotype acute leukemia, T-myeloid, NOS
Provisional entity: natural killer (NK) cell lymphoblastic leukemia/lymphoma
Immunophenotype Analysis in AML
In less than 20 % of cases of AML, morphology and cytochemistry are inconclusive or insufficient to distinguish AML and ALL [68]. By using a combination of monoclonal antibodies recognizing B-cell, T-cell, and myeloid antigens, it is possible to diagnose most of these difficult cases [69]. Confirmation of the rare cases of M0 AML in children and adults requires immunophenotyping or flow cytometric or ultrastructural peroxidase studies. Adult AML-M0 has been associated with an immature myeloid profile (CD34+, terminal deoxynucleotidyl transferase (Tdt)+, CD13+, and CD33+), in contrast to pediatric AML-M0, which usually lacks Tdt or CD34 but expresses bright CD33 with weak or negative CD13 [70]. Coexpression of CD7 is observed in both adult and pediatric M0 cases. The M7 FAB subtype also may be suspected based on morphology or histochemistry, but it needs to be confirmed by either ultrastructural histochemistry (platelet peroxidase) or immunophenotypic analysis (identification of platelet or megakaryocytic antigens) [62]. Flow cytometric analyses in AML can also be useful for detecting MRD after induction or consolidation chemotherapy [2].
Bilineal and Biphenotypic Leukemias
Approximately 5–15 % of cases of acute leukemia have morphologic, cytochemical, immunophenotypic, or genetic evidence of more than one hematopoietic lineage [71–73]. In these hybrid or acute mixed-lineage leukemias, individual blasts may express features of more than one lineage (biphenotypic leukemia), or there may be two distinct populations of blasts (bilineal leukemia). In some cases leukemia may undergo a switch in immunophenotype with therapy. The latest revision of the WHO classification of hematopoietic neoplasms includes a separate category of “acute leukemias of ambiguous lineage” that includes subgroups of acute undifferentiated leukemia and those with either BCR-ABL, MLL rearrangements, B/myeloid, and T/myeloid subgroups [66]. The pathogenesis of these hybrid leukemias remains poorly understood and no cytogenetic or molecular abnormalities specific to this group of leukemias have been found. Some molecular abnormalities seen in this group include BCR-ABL and MLL rearrangements [66]. In a study from St. Jude Children’s Research Hospital, the overall survival of both B/myeloid and T/myeloid subgroups was similar to AML but inferior to children with ALL. Based on their experience they recommended treating these patients initially with AML-directed therapy, but switching to lymphoid-directed therapy for those with a poor response, as these patients often responded to therapies including vincristine, prednisone, and l-asparaginase [74]. There is some controversy over these recommendations as a German study by Gerr et al. [75] found better outcomes in B/myeloid cases treated with ALL-directed therapy compared with those treated with AML-directed therapy or combined therapy. They recommended treating patients expressing the B-cell antigens cytCD79a or cytCD22, lymphoblastic morphology, and those with ETV6/RUNX1 with ALL-directed therapy. It should be mentioned that their patients were diagnosed with biphenotypic leukemia using pre-2008 WHO criteria. None of the subgroup of children with ETV6/RUNX1 was MPO positive or had sufficient evidence of monocytic differentiation, and thus, would not meet current WHO criteria for acute leukemias of ambiguous lineage.
Chromosomal and Genetic Changes
Sixty percent of children with AML have one of several specific chromosomal translocations involving either the AML1/CBFB core binding factor complex—t(8;21), inv(16), or t(16;16); the retinoic acid receptor—t(15;17); or the MLL protein (rearrangements of 11q23). These translocations create specific fusion transcripts (AML1-ETO, PML-RARA, CBFB-MYH11, and MLL fusions). The resultant chimeric transcription factors can confer increased self-renewal and interfere with the differentiation of myeloid progenitors, with different translocations correlating with distinct morphological FAB subgroups. For instance, children with PML-RARA expression resulting from t(15;17) have acute promyelocytic leukemia (FAB M3). All cases of FAB M4eo, a subtype of AML with marked eosinophilia, have inv(16) or t(16;16). t(8;21) is often associated with FAB M2 and MLL (All-1, HRX) rearrangements with the FAB M4 and FAB M5 subtypes [65, 76, 77]. Distinct MLL rearrangements can result in either ALL or AML. In contrast to the more common t(9;11), translocation in AML creating the MLL-AF9 fusion gene, the majority of infant precursor B-ALL is the result of a t(4;11) translocation creating the MLL-AF4 fusion gene [78].
These lesions are thought to represent important initiating events in the pathogenesis of AML; however, these lesions are apparently insufficient for leukemogenesis and require additional cooperating mutations for the development of AML. Evidence of the multistep pathogenesis of AML comes from the demonstration that certain fusion transcripts such as AML1-ETO are apparently insufficient for leukemogenesis, as demonstrated by the mouse models [79–82], the presence of fusion gene transcripts in patients with long-term remission [83–85], and the finding of such transcripts in blood collected from newborns who did not develop t(8;21) AML for up to 10 years [86].
Gilliland and colleagues have proposed a model in which AML develops from a combination of the earlier described “Class II” mutations, which primarily cause a differentiation arrest and increased self-renewal, with “Class I” mutations that cause increased proliferation and survival [87]. Such class I mutations include activating mutations in N-RAS or K-RAS as well as constitutive activation of receptor or cytoplasmic tyrosine kinases such as FLT3 and C-KIT. Reported frequencies of RAS mutations in AML range from 25 to 44 % [88]. Activating mutation in FLT3 is the most common mutation in AML seen in 30–35 % of adults [87] and 12–25 % of pediatric and young adults with AML.
Approximately 20 % of children with AML do not have clonal chromosomal abnormalities identified by routine cytogenetics [51]. An extended spectrum of chromosomal alterations can be appreciated using comparative genome hybridization (or array-based CGH), single nucleotide polymorphism arrays [89], or large-scale DNA methylation profiling [90]. Mutations in a variety of single genes are involved in the pathogenesis of AML some of which affect the prognosis of the disease as described later in this article. These include mutations of FLT3, c-KIT, NPM1, RAS, PTPN11, TIF1, CEBPA, and PU.1. The advent of next generation genome sequencing is now allowing a better understanding of the complement of cooperating mutations in AML [91].
Clinical and Laboratory Manifestations
The signs and symptoms of the child presenting with AML reflect the degree of bone marrow infiltration with leukemic cells as well as the extent of extramedullary spread. Acute myelogenous leukemia is rarely diagnosed as an incidental finding on a routine blood count in an asymptomatic child. Most children present with pallor, fatigue, bleeding, or fever as manifestations of their underlying anemia, thrombocytopenia, and neutropenia (Table 21.2) [92]. Lymphadenopathy, bone pain, and arthralgias are presenting features in less than 20 % of children with AML and are more commonly observed in children with ALL [93]. Approximately 50 % of children with AML have hepatomegaly or splenomegaly (more than 5 cm below the costal margin in 10–30 % of cases). Infants with AML are more likely to have organomegaly, leukemic cutis, CNS leukemia, and high presenting leukocyte counts as compared with older children with AML [65].
Table 21.2
Presenting clinical and laboratory features in children with AML (AML-BFM-83 Study)
Feature | % of Patients |
|---|---|
Sex (M/F) | 55/45 |
Age (year) | |
<2 | 18 |
2–9 | 39 |
= 10 | 43 |
Leukocyte count (×109/L) | |
<20 | 48 |
20–100 | 35 |
>100 | 17 |
Hemoglobin | |
=8 g/dL | 46 |
>8 g/dL | 51 |
Platelets (×109/L) | |
=50 | 53 |
>50 | 47 |
Liver (cm) | |
=5 | 86 |
>5 | 14 |
Spleen (cm) | |
=5 | 87 |
>5 | 12 |
CNS | |
Absent | 88 |
Present | 5 |
The anemia associated with both ALL and AML is usually normocytic, with occasional teardrop and nucleated red blood cells observed on peripheral smear. Initial hemoglobin levels ranged from 2.7 to 14.3 g/dL (median, 7 g/dL) in one large series of children with AML [93].
Bleeding in a child with newly diagnosed AML is usually due to thrombocytopenia with or without disseminated intravascular coagulation. As in adults, most thrombocytopenic bleeding occurs when the platelet count is less than 20 × 109/L. Approximately 50 % of children with AML present with platelet counts of less than 50 × 109/L. Disseminated intravascular coagulation is usually associated with acute promyelocytic leukemia (FAB M3), but the FAB M4 and M5 subtypes of AML in children have also been associated with coagulation abnormalities and early death from hemorrhagic complications [94].
Initial leukocyte counts in most children with AML are less than 50 × 109/L. However, 20 % will have white blood cell counts greater than 100 × 109/L at diagnosis (Table 21.2). Leukostasis or intravascular clumping of blasts leading to hemorrhage and infarction can lead to life-threatening complications in children with AML, especially when the leukocyte count is in excess of 200 × 109/L [95]. Leukostasis often manifests as somnolence, seizures, stroke, or tachypnea with hypoxemia.
Differential Diagnosis
A careful history and physical examination, as well as an examination of peripheral blood and bone marrow, result in a straightforward diagnosis of AML in approximately 90 % of cases. However, for the difficult cases other diagnoses must be systemically excluded. Some of these include chronic myeloproliferative disorders, myelodysplastic syndromes, aplastic anemia, and overwhelming infections that result in leukemoid reactions or neutropenia.
As previously mentioned, newborns with DS or trisomy 21 mosaicism may show a transient proliferation of blast cells [16, 23, 96]. This so-called transient myeloproliferative syndrome is often clinically and hematologically indistinguishable from congenital AML. Expectant management when clinically appropriate may allow clarification, as can genetic testing for GATA1 mutations [22].
Extramedullary Leukemia
Infants with AML have a propensity for extramedullary dissemination of disease. In a review of AML in 29 infants at St. Jude Children’s Research Hospital, 13 were found to have leukemia cutis, and 11 presented with CNS leukemia [97]. The subcutaneous chloromas and leukemia cutis are bluish to slate gray in color, may appear in all sites, and are palpated as rubbery nodules of the deep skin. It has been postulated that a more intense blood supply present in neonatal skin and the propensity for monocytes to migrate to extramedullary sites may account in part for the high frequency of leukemia cutis seen in neonates and infants with AML.
Approximately 5–15 % of children with AML have blasts detected in cerebrospinal fluid (CSF) at diagnosis [98, 99], but most of these children are asymptomatic. The clinical manifestations of CNS leukemia reflect leukemic infiltration of the leptomeninges and can include signs and symptoms of increased intracranial pressure, as well as isolated cranial nerve palsies (most common is palsy of the cranial nerve VII). Testicular involvement is extraordinarily rare in children with AML and, like CNS leukemia, is associated with the M4 and M5 FAB subtypes [100].
Chloromas or myeloblastomas are seen in fewer than 5 % of children with AML [8, 100]. As in adults, chloromas may appear simultaneously with bone marrow infiltration or may present as the initial clinical manifestation of leukemia weeks to months before overt bone marrow disease can be detected. Chloromas in children are frequently seen in the bones and soft tissues of the head and neck (often orbits), skin, and epidural areas [101].
Treatment
The prognosis for children with AML has gradually improved during the past 25 years [99, 102–108]. Improvements have been made as a result of intensification of treatment, improved supportive care, and risk-stratified treatments [3]. The likelihood of achieving a complete remission (CR) is about 80–90 % with 5-year overall survival rates now approaching 60 % [3]. Most children with newly diagnosed or suspected leukemia are referred to a tertiary care center staffed by pediatric hematologists–oncologists. The majority of children with AML are treated according to cooperative group clinical trials, such as those by the Children’s Oncology Group (COG) [a merger of the Pediatric Oncology Group (POG), and Children’s Cancer Group (CCG)], Berlin–Frankfurt–Münster (BFM) Group, and the UK Medical Research Council (MRC) [102, 104, 106, 107].
During the immediate hours after diagnosis, prompt recognition and treatment of life-threatening complications is necessary. The most likely oncologic emergencies in this setting include leukostasis, sepsis, hemorrhage, and mass effect from myeloblastomas (e.g., spinal cord compression).
In one large series of children with AML, 10 % of patients died as a result of hemorrhage, leukostasis, or both before or during the first 12 days of therapy [94, 109]. Most of these early deaths were due to intracerebral hemorrhage, and the major risk factors were hyperleukocytosis, coagulopathy, and acute monocytic leukemia. Hyperleukocytosis was defined as a white blood cell count greater than 100 × 109/L. Leukapheresis (or exchange transfusion in very young infants) is well tolerated and very effective in transiently lowering the white blood cell count [110]. Hydroxyurea or the initiation of induction chemotherapy is often needed in this setting after vigorous hydration, alkalinization of the urine with intravenous sodium bicarbonate, and administration of allopurinol [111]. Hyperuricemia, when significant, can be very effectively treated with rasburicase, a recombinant urate oxidase [112]. The tumor lysis syndrome (hyperkalemia, hyperphosphatemia, hypocalcemia, and hyperuricemia) is more common in children with ALL than in those with AML [95].
Remission Induction
Once the patient is stabilized, remission induction therapy should begin without delay. Most pediatric AML remission induction protocols employ a 7- to 10-day course of cytosine arabinoside (ara-C), plus 2 to 3 days of doxorubicin, daunorubicin, or mitoxantrone, with or without thioguanine or etoposide [113–118]. Approximately 80–90 % of children with AML achieve CR using current protocols. For children under 1 year of age or who have a body surface area of less than 0.6 m2, drug doses should be calculated on a milligram per kilogram (mg/kg) basis, rather than on a milligram per square meter (mg/m2) basis, to avoid excessive toxicity.
Because of the narrow therapeutic index for ara-C and the anthracyclines, one can expect 2–3 weeks of pancytopenia before there is evidence of bone marrow recovery. Therefore, the remission induction period is associated with considerable morbidity and mortality [117, 118]. Approximately 3–10 % of children die during induction therapy as a result of direct leukemic causes, infection, or hemorrhage [104–107, 119].
Chemotherapy-induced gastrointestinal toxicity contributes to the risk of bacterial or fungal sepsis [120, 121]. The most severe manifestation of gastrointestinal toxicity has been referred to as enterocolitis or typhilitis [122]. Severe neutropenia coupled with chemotherapy-induced mucosal damage to the distal ileum and proximal colon is thought to be a predisposing factor. Typhlitis is characterized by right lower quadrant abdominal pain with or without rebound tenderness. In severe cases, there may be vomiting, ileus, abdominal distention, fever, and septic shock. A relative lack of gas from the right lower quadrant and minimal distention of the small bowel may be seen on plain abdominal radiographs. Abdominal ultrasound and CT scans may reveal thickening of the wall of the cecum. The management of a child with typhlitis should include bowel rest, intravenous fluids, broad-spectrum antibiotics, and appropriate blood components. Surgery is reserved for intestinal perforation, abdominal wall fasciitis, or massive bleeding. In the differential diagnosis of typhlitis, one must also consider common surgical conditions of childhood, including appendicitis or intussusception. Typhlitis is less common with remission induction regimens using daunorubicin rather than doxorubicin combined with ara-C [123].
Therapy for AML in Down Syndrome
Down syndrome children with AML manifest an increased sensitivity to chemotherapy and as a group have had increased treatment-related mortality with approaches using intensive timed induction therapy, higher doses of anthracyclines, or allogeneic bone marrow transplantation (allo-BMT) [107, 124]. Recognition of this sensitivity has led to high rates of cure with this group using standard or reduced therapy [106, 107, 125–127]. The greatest risk for AML in Down syndrome children is in the first years of life, but when it does occur in older children (>4 years of age) the risk of relapse can be quite high (6 year EFS of 28 %) with less intensive therapy [128]. Tailored intensive therapy with improved supportive care appears to benefit this older group [129].
Central Nervous System Prophylaxis
In contrast to the clear benefits of improved long-term DFS with CNS-directed prophylaxis in ALL [2], the benefit of CNS-directed therapy in AML is less clear. Without CNS-directed therapy, the incidence of isolated CNS relapse during hematologic remission in children with AML is approximately 20 % [98, 99]. Children with the M4 and M5 subtypes of AML have the highest risk of CNS relapse [130]. Intrathecal methotrexate [131] or cytosine arabinoside [116] or cranial irradiation plus intrathecal methotrexate [114] have all been used to reduce the risk of CNS relapse in children with AML. However, it has yet to be demonstrated in a prospective randomized, clinical trial that CNS prophylaxis improves disease-free survival in children with AML. The only data supporting the use of CNS prophylaxis in AML derive from BFM protocols [114, 132]. The French Cooperative group has used CNS prophylaxis primarily in patients with M4 or M5 subtypes and those with a WBC count greater than 50 × 109/L [105]. While the majority of pediatric oncology groups currently use intrathecal CNS prophylaxis, few adult centers do.
Cranial radiation is not generally used for CNS prophylaxis. Although the German BFM group found an increased risk of relapse in CNS negative patients when CNS radiation was omitted, this was only in a group of nonrandomized patients [132]. This study was also performed in an era of less intensive and effective regimens, and has not been verified by other groups. This could reflect the effective CNS penetration of high-dose ara-C. Based on these results and to avoid long-term sequelae of irradiation prophylactic cranial irradiation is no longer being used in BFM trials [133].
About 10–15 % of children with AML have blasts in the CSF at diagnosis. Interestingly, this finding does not appear to have an adverse impact on overall survival [99, 103, 134]. Overt CNS leukemia has been treated with weekly intrathecal methotrexate or ara-C until the CSF is clear of blasts. Cranial irradiation may be given in those patients who are unable to clear their CSF blasts with intrathecal and systemic therapy.
Postremission Therapy
Successful remission induction therapy reduces the leukemic burden by two to four orders of magnitude but does not eradicate disease. Therefore, additional therapy is necessary to achieve prolonged remission durations in both children and adults with AML. The intensity and duration of post-remission chemotherapy, as well as the role of bone marrow transplantation in first remission, remain controversial. Pediatric AML studies initiated during the 1970s and early 1980s tested whether modestly myelosuppressive combination (maintenance) chemotherapy [135, 136], nonspecific immunotherapeutic agents (e.g., bacillus Calmette–Guérin [BCG]) [137], or splenectomy [138] would prolong remission duration. Disease-free survival rates with these approaches were less than 30 % at 5 years, and there was no obvious benefit for splenectomy or adjuvant BCG.
Because of these disappointing results, other treatment approaches were explored, including intensification/consolidation chemotherapy and allo-BMT early in first remission [99, 139, 140]. Intensification chemotherapy was based on the steep dose–response curve for many of the chemotherapeutic agents such as ara-C. Other strategies included the use of sequential non-cross-resistant drug combinations in an effort to circumvent the problem of acquired drug resistance [99]. Although these strategies were explored in nonrandomized clinical trials, the overall results suggested a benefit for this approach.
During the 1980s, it became apparent that high doses of ara-C could produce remissions in patients with AML who were refractory to conventional-dose ara-C [141]. These exciting results led to the use of high-dose ara-C as consolidation therapy during first remission [131]. An important Cancer and Leukemia Group B (CALGB) protocol for adults with AML randomly assigned patients to receive four courses of ara-C at one of three doses: 100 mg/m2 for 5 days, 400 mg/m2 for 5 days, or 3 g/m2 on days 1, 3, and 5. The probability of remaining in remission for patients under 60 years of age was 44 % in the 3 g/m2 group, which was significantly different from the 24 % for the 100 mg/m2 group and 29 % for the 400 mg/m2 group [142]. Based on these results as well as the previously mentioned studies, one or more courses of high-dose ara-C were included in the consolidation phase of many pediatric AML protocols. Leukemias involving rearrangements of the core binding factor genes, namely t(8;21), inv(16), or t(16;16), appear to respond especially well to repetitive cycles of high-dose cytarabine [143, 144].
Stem Cell Transplantation
The decision whether to perform a stem cell transplant (SCT) in first clinical remission (CR1) remains controversial in pediatric AML. Data from several but not all pediatric AML clinical trials show a disease-free survival advantage for allo-BMT from an HLA-matched family member as compared with chemotherapy [113, 116, 118].
During the mid-1970s, allo-BMTs from histocompatible family donors were first evaluated in children and young adults with AML in first remission [139, 140]. About 25 % of potential transplant candidates were eligible by virtue of having an HLA-identical family donor. The early data were quite encouraging, with 5-year disease-free survival rates of 50–70 % compared with less than 30 % for chemotherapy. These results were also obtained in a very selected group of patients. The relapse rate of AML after allo-BMT was lower than that after chemotherapy (20 % versus 60 %), but the difference in leukemia-free survival was less dramatic because of fatal post-BMT complications from graft-versus-host-disease and infections from immunosuppression.
The CCG was one of the first groups to prospectively compare the outcome of chemotherapy and allo-BMT and avoid the bias of patient selection [146]. When the data were analyzed on the basis of intent to transplant (HLA-identical donor available), a statistically significant disease-free survival advantage was found for transplant compared with chemotherapy (48 % versus 36 %).
Similar results were reported in other studies [113, 114, 116–118]. However, with continued improvement in the outcome after intensive chemotherapy, other studies have not demonstrated a statistically significant disease-free or overall survival advantage for allo-BMT as compared with chemotherapy [117]. Post-hoc subset analysis suggests that distinct molecular and prognostic subgroups might benefit from different treatment strategies [147]. Nonetheless, MRC studies have not demonstrated a statistically significant survival advantage of even high-risk patients for allo-BMT in first CR [104].
Methodological inadequacies and limited patient numbers prevent an uncontroversial interpretation of published studies [148]. However, there is currently a consensus even within COG against transplanting children in first CR with favorable risk groups such as t(8;21), inv(16), APL, and Down syndrome related AML.
Because of the promising results of autologous marrow transplantation (ABMT) in patients with AML in second remission [149], randomized clinical trials comparing ABMT with chemotherapy in children with AML in first remission were initiated [113, 116, 117]. In several studies, marrow was purged with either 4-hydroperoxycyclophosphamide or mafosfamide. The results of the ltalian [113], Pediatric Oncology Group [113], Children’s Cancer Group [118], and MRC [104] studies have not shown a disease-free survival advantage for ABMT over chemotherapy.
Acute Promyelocytic Leukemia
The biology and molecular genetics of acute promyelocytic leukemia (APL, FAB M3) are identical in children and adults [109, 150]. Acute promyelocytic leukemia is commonly associated with life-threatening bleeding and coagulopathy; thus initiation of therapy should proceed immediately based on morphological and cytochemical evaluation of blood or bone marrow smears, without waiting for molecular confirmation [151]. Routine lumbar puncture should be delayed until resolution of any coagulopathy. Aggressive supportive therapy is necessary for the coagulopathy including maintaining a platelet count greater than 30–50 × 109/L and transfusing fresh frozen plasma and/or cryoprecipitate to maintain fibrinogen greater than 100–150 mg/dL during the first ten days of induction therapy or resolution of the coagulopathy [152, 153]. The use of all-trans-retinoic acid (ATRA) is unique to this type of AML and induces differentiation of APL blasts, lessening the coagulopathy. In standard risk disease (WBC <10 × 109/L), chemotherapy is delayed for 1–3 days, but for high-risk disease (WBC >10 × 109/L), chemotherapy is started concurrently since ATRA can induce a hyperleukocytosis with initial worsening of the coagulopathy [152]. Leukopheresis, sometimes performed to reduce a high WBC, is generally avoided as the resultant cell trauma may release procoagulants, worsening the coagulopathy.
The introduction of ATRA which specifically targets the PML-RARA fusion gene underlying APL has revolutionized the treatment of this subtype of AML, transforming a once mostly fatal disease into one of the most curable malignancies [154]. The use of ATRA in APL represents one of the first examples of molecularly targeted therapy. However, the use of ATRA is associated with a respiratory distress/capillary leak syndrome, also known as the APL differentiation syndrome or ATRA syndrome, that can manifest as fever, rash, pulmonary infiltrates, pleural and pericardial effusions, respiratory distress with hypoxemia, congestive heart failure, and can progress to cardiopulmonary failure [153]. Prompt administration of dexamethasone is indicated if this syndrome is suspected, with the temporary cessation of ATRA in severe cases, or those not responding to steroids [151]. Different groups have advocated prophylactic administration of dexamethasone for an elevated WBC greater than 10 × 109/L or 50 × 109/L [152, 155]. Concurrent treatment with chemotherapy has dramatically lessened the incidence of ATRA syndrome. Pseudotumor cerebri can also be seen, and is more common in children than adults, but usually abates within a week after withdrawal of ATRA. The drug can usually be safely restarted at a reduced dose after these side effects.
The standard APL induction therapy in adults combines ATRA with an anthracycline, most commonly idarubicin. Consolidation with at least two rounds of anthracycline-based chemotherapy with ATRA is then generally performed. The addition of ara-C in induction and consolidation for high-risk patients with WBC greater than 10 × 109/L has resulted in improved outcome in adults [156]. Most groups use maintenance therapy for 1–2 years using ATRA and low-dose anti-metabolite therapy. A beneficial role of maintenance therapy in APL has been demonstrated by several randomized trials [157, 158]; however, there may not be a benefit of maintenance therapy in those in which a molecular remission is achieved after intensive consolidation therapy [159].
Arsenic trioxide has proved to be one of the most effective single agents in APL in inducing remission in both children and adults with relapsed APL [160, 161]. This has led to trials incorporating arsenic in upfront therapy. The addition of arsenic to consolidation in a recent North American Intergroup trial led to significant improvements in EFS and OS [162]. In an effort to reduce the total anthracycline dose and risk of cardiotoxicity but maintain comparable outcomes a current COG trial utilizes high-dose ara-C in consolidation for even standard risk children and also incorporates arsenic trioxide in consolidation. The effectiveness of ATRA and arsenic has even led to trials that have successfully treated APL without the use of conventional chemotherapy using only these two agents with the addition of gemtuzumab ozogamicin (Myelotarg) for high-risk patients [163]. Such a strategy would be particularly attractive in younger children to reduce the long-term side effects of conventional chemotherapy.
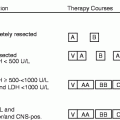
Prognostic Factors (Table 21.3)
Table 21.3
Prognostic factors in childhood AML
Good prognostic features |
Down syndrome AML <4 years of age |
Favorable cytogenetics |
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 (AML1-ETO) |
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 |
APL with t(15;17)(q22;q12); PML-RARA |
Normal karyotype with: |
NPM1 without FLT3-ITD |
CEBPA mutations |
Adverse prognostic features |
WBC > 100,000 cells/μL |
Secondary AML |
Unfavorable cytogenetics |
Abn(3q) |
−5/del(5q) |
−7 |
Complex karyotype |
Unfavorable molecular abnormalities |
FLT3-ITD |
IDH1, IDH2 mutations |
Slow response to induction therapy |
BM blast >5 % day 15 |
The response to induction therapy is highly predictive of outcome. Slow response as measured by a bone marrow blast percentage >5 % on day 15 has been associated with worse outcomes in CCG and BFM trials [107, 164]. Similarly, the early response to therapy after 10 days of therapy was highly predictive of outcome in the MRC10 trial [165]. The presence of residual leukemia cells at the end of induction below the limits of detection by morphological evaluation but measurable by multiparameter flow cytometry or RT-PCR, termed MRD, has also been shown to carry a worse prognosis [166, 167]. However, in multivariate analysis, including FAB classification and karyotype, MRD did not prove to be of independent prognostic significance in a BFM trial [167]. Further characterization of the significance of MRD is being examined in COG trials AAML03P1 and AAML0531. It does appear that in APL MRD monitoring by RT-PCR for PML-RARA transcripts in bone marrow samples obtained after consolidation, during maintenance or after therapy, is predictive of impending relapse [168–170].
A variety of pretreatment-related factors have been shown to be of prognostic significance in AML [171, 172]. A high WBC >100 × 109/L has been associated with a lower CR and EFS rate [107, 134] as well as an increased risk of relapse in some [173, 174] but not all studies [114, 118]. Being either underweight or overweight increased the risk of treatment-related mortality [175]. African-American and Hispanic ethnicity was associated with a worse survival as compared with Caucasian children with AML in an analysis of over 1,600 patients treated on CCG trials [176]. Increasing age has also been associated with worse survival in both adults and children [177, 178]. As mentioned previously, AML in Down syndrome, especially when occurring in the first few years of life has a high cure rate [106, 107, 125–127].
Although these pretreatment variables have prognostic significance, with the exception of AML in Down syndrome, it is primarily the diagnostic karyotype that has proved most useful for treatment stratification [179]. Clonal chromosomal abnormalities identified at diagnosis by cytogenetic analysis have proved to be one of the strongest determinants of outcome [51, 180]. Three cytogenetic risk groups have been identified: low (favorable), intermediate, and high (adverse) risk. There is general consensus that patients with t(15;17), inv(16)/t(16;16), or t(8;21) are of low risk and that patients with monosomy 5 or 7, del(5q), complex karyotypes (defined variably as >3 or >5 unrelated abnormalities), or abnormalities of 3q26 are at high risk [103, 179, 180]. Patients with other abnormalities and a normal karyotype have been grouped into an intermediate risk group, although the risk for specific defects within this category is being refined [181].
In addition to these clonal chromosomal abnormalities, specific molecular abnormalities not evident by cytogenetic analysis also have important prognostic significance. These include activating mutations in receptor tyrosine kinase genes, primarily FLT3 and c-KIT. Flt3 can be activated by either and internal tandem duplication, FLT3-ITD, or an activating loop mutation, FLT3-ALM. The presence of FLT3-ITD carries a worse prognosis [171]. Further analysis has revealed variation in the mutant to wild-type allelic ratio in different AML samples, and it appears to be primarily those with an allelic ratio of greater than 0.4 who manifest a particularly worse prognosis. This group had a relapse rate of nearly 80 % and a survival rate of <20 % [182]. Additional studies indicate that it is the presence of FLT3-ITD in the CD34+/CD33− subpopulation of bone marrow cells rather than confinement to more mature CD34+/CD33+ cells that confers a worse prognosis [183]. Activating mutations in the c-Kit receptor tyrosine kinase have been found in 3–11 % of pediatric AML, with a higher frequency of nearly 40 % in CBF leukemias [184]. Several studies have demonstrated a worse prognosis with c-KIT mutations in adult AML [185–187], while results in pediatric AML have been mixed [184, 188, 189]. Mutations in the nucleophosmin member 1 (NPM1) are seen in about 30 % of adult AML samples [190] but only about 8 % in children AML [191]. The incidence is significantly higher in normal karyotype AML, a group in which its presence, in the absence of FLT3-ITD mutations, confers an improved prognosis, comparable to those in the favorable cytogenetic risk group [190, 191]. Mutations in CEBPA have been associated with a favorable outcome in adult AML [192] although it is less common in pediatric AML and its significance less clear.
Cytogenetically normal AML (CN-AML) is considered to have intermediate risk, but represents the largest karyotypic group. The identification of other molecular abnormalities within CN-AML may identify subgroups that might benefit from more intensive or novel therapies. For instance, the presence of the constitutively activated FLT3-ALM mutation did not appear to carry a worse prognosis in several studies [189, 193], but was associated with worse survival in the subgroup of young adults with cytogenetically normal AML lacking FLT3-ITD [194]. Other abnormalities identified in adults with CN-AML which confer a worse prognosis include IDH1 and IDH2 mutations [195] and Wilms Tumor 1 gene mutations [196]. The usefulness of these observations in childhood AML, however, may be limited as one recent study failed to detect IDH1 mutations in 257 pediatric AML samples [197]. Efforts are also under way to identify gene-expression and microRNA-expression profiles to predict outcome in CN-AM [198–201]. Models have attempted to use molecular analyses to stratify prognosis in CN-AML [202].
Management of Relapse and Refractory Disease
Management of children with relapsed or refractory AML is a major challenge. For those that do relapse the duration of the first remission has a major impact on outcome. Children who relapse early (defined as remission less than 1 or 1.5 years in different studies) fare poorly with long-term survival rates of 10–24 % as compared with 40–54 % for late relapses [203–205].
A variety of different reinduction regimens have been used in children with refractory or relapsed AML, including high-dose ara-C with or without l-asparaginase [206], mitoxantrone [207], or fludarabine and G-CSF [208]. Other regimens include amsacrine and etoposide (VP-16), with or without azacytidine [209], etoposide and mitoxantrone with cyclosporine [210], and 2-chlorodeoxyadenosine [211]. Clofarabine has recently emerged as highly effective single drug in relapsed ALL [212] and also has activity as a single agent in relapsed and refractory pediatric AML [213]. Additional trials using clofarabine in combination therapy such as with etoposide and cyclophosphamide appear to hold some promise [214]. Mylotarg also appears to have efficacy in this setting for CD33+ AML with a 30 % response rate in recurrent pediatric AML as a single agent [215–217]. Like clofarabine it may prove more useful in combination with other agents.
The second CR rates are about 20–50 % with the earlier discussed regimens. Unfortunately, the durations of second remission are usually brief, especially for patients with short first remissions. For these reasons, bone marrow transplantation is recommended for patients who achieve a second remission [218]. Five-year DFS rates after allogeneic transplantation for children with AML in CR2 have improved to 45–58 % [219, 220] with potentially improved survival after late relapse [221]. Transplant results for those relapsed patients not in remission are much lower, e.g., 20 % 5-year DFS [220].
Transplant related mortality (TRM) in this setting is significant and appears related to age at transplant, with early TRM (by day 100) reported in the ≤10-year-old population of 14 % and for older children (age 11–18) 25 %. Late TRM (5 year) rates for these populations have been reported as 19 % and 41 %, respectively [220].
New Therapies
It is unlikely that further intensification or improvements in supportive care will significantly improve outcomes of AML therapy. Further substantial improvements in outcome are more likely to come from the identification of new active agents or therapeutic strategies and the development of tailored targeted therapies for defined molecular subgroups.
Cellular therapy using haploidentical-related-donor natural killer cell infusions represents one such promising new strategy that has minimal toxicity [222]. This therapy is currently being trialed in COG for relapsed and refractory AML in children.
A variety of small molecule inhibitors of receptor tyrosine kinases primarily targeting FLT-3 have been under evaluation in children and adults [223, 224]. These inhibitors as single agents have been able to induce partial to complete remission, even in chemotherapy-resistant disease, but, in general, these remissions have not been sustained. Two of these, lestaurtinib (CEP-701, Cephalon) and midostaurin (PKC412, Novartis), inhibit multiple tyrosine kinases, are orally available, and have reached phase III clinical trials [225, 226]. Currently, their usefulness in combination with chemotherapy is being further examined.
Other classes of targeted agents under active investigation include farnesyl transferase inhibitors such as Tipifarnib (Zarnestra, R115777, Johnson & Johnson) which is also orally available [227], proteosome inhibitors such as bortezomib (Velcade, PS341, Millennium Pharmaceuticals) [228], mammalian target of rapamycin (mTOR) inhibitors such as rapamycin (Sirolimus, Rapamune, Wyeth [229]), histone deacetylase inhibitors such as Vorinostat (SAHA, Patheon, Inc.) or valproic acid [230, 231], and DNA methylase transferase inhibitors such as decitabine (Dacogen, aza-C, MGI Pharma) [232].
Supportive Care
General principles of supportive care in children with AML include replenishment of deficient blood components; prompt recognition and treatment of metabolic, nutritional, and infectious complications; placement of central venous access devices; and psychosocial intervention and support.
Relapsed leukemia remains the primary cause of death in children with AML, but infections continue to cause morbidity and mortality. Susceptibility to bacterial and fungal infection is the result of neutropenia caused initially by bone marrow replacement with leukemia and later by chemotherapy-induced bone marrow suppression, cellular and humoral immunosuppression, breakdown of skin and gastrointestinal barriers from therapy, and the use of central venous access devices [120, 121]. Children receiving high-dose ara-C are at particular risk for alpha-hemolytic streptococcal infection [233] and in the setting of fever after ara-c empiric therapy including vancomycin is recommended. As the intensity of AML therapy has increased, so has the rate of invasive fungal disease.
The empirical use of broad-spectrum antibiotics and amphotericin B or fluconazole in febrile neutropenic patients, the prophylactic use of trimethoprim–sulfamethoxazole to prevent Pneumocystis carinii pneumonia, and the use of acyclovir in children with herpes zoster virus or varicella zoster virus infections have contributed significantly to the improved outlook in childhood acute leukemia [120, 121]. Oral nonabsorbable antibiotics and protected environments (i.e., laminar airflow) have not been advocated. The use of hematopoietic growth factors (e.g., granulocyte- and granulocyte-macrophage-colony-stimulating factors [G-CSF and GM-CSF]) has not been shown to increase remission rates or overall survival in children with AML [118].
Hemorrhagic deaths have generally been preventable with the liberal use of platelet concentrates, particularly in patients with platelet counts below 10–20,000 per cubic mm. Alloimmunization to random platelet concentrates is a potential problem, especially for children with AML who require frequent blood component support. HLA-matched platelets may be effective when patients become refractory to random donor platelet concentrates. All blood products should be leukodepleted and irradiated [234].
Consequences of Survival
With an increasing number of children with AML surviving disease-free for long periods, attention has been drawn to late effects of leukemia and its treatment, including the risk of second neoplasms [235, 236]. Growth and development and sexual maturation have generally been normal in both boys and girls who have completed chemotherapy for AML, but the reproductive capacity of successfully treated patients’ needs to be established. Gonadal failure, short stature, and secondary malignancies are more commonly observed after BMT [237]. Late cardiotoxic effects of anthracyclines are a potential problem, and children should be monitored with echocardiograms on a regular basis (e.g., every other year) [238]. A high rate of cardiomyopathy primarily in DS AML children was seen in an earlier POG trial that utilized a particularly high dose of anthracyclines [239]. This higher rate has not been seen in other protocols [240], but does point to an increased sensitivity in this patient population that needs to be considered in study design. Current studies are addressing quality-of-life issues, as they will undoubtedly have an impact on the selection of treatment options (e.g., marrow transplantation versus chemotherapy in first remission) [241].
For an increasing proportion of children with AML, their disease has become a long-term problem. Patients and their families must be counseled in these terms. The effects of leukemia on parents and siblings of the affected child have taught us that the impact of this disease is shared by the entire family.
References
1.
American Cancer Society. Cancer facts and figures in 2009. Atlanta: American Cancer Society; 2009.
3.
4.
5.
Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519.PubMed
6.
Smith MA, Ries LAG, Gurney JG, Ross JA. Incidence and survival among children and adolescents: United States, SEER Program 1975–1995. In: Ries LAG, Smith MA, Gurney JG, et al., editors. NIH Pub No 99–4649. Besthesda, MD: National Cancer Institute, SEER Program; 1999. pp. 17–34.
7.
12.
Richkind KE, Loew T, Meisner L, Harris C, Wason D. Identical cytogenetic clones and clonal evolution in pediatric monozygotic twins with acute myeloid leukemia: presymptomatic disease detection by interphase fluorescence in situ hybridization and review of the literature. J Pediatr Hematol Oncol. 1998;20:264–7.PubMedCrossRef
13.
14.
Jongmans MC, Kuiper RP, Carmichael CL, et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: clues for improved identification of the FPD/AML syndrome. Leukemia. 2010;24:242–6.PubMedCrossRef
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


