B lymphoblastic leukemia
B lymphoblastic leukemia, not otherwise specified
B lymphoblastic leukemia with recurrent genetic abnormalities
B lymphoblastic leukemia with t(9;22)(q34;q11.2); BCR-ABL1
B lymphoblastic leukemia with t(v;11q23); MLL rearranged
B lymphoblastic leukemia with t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1)
B lymphoblastic leukemia with hyperdiploidy
B lymphoblastic leukemia with hypodiploidy
B lymphoblastic leukemia with t(5;14)(q31;q32); IL3-IGH
B lymphoblastic leukemia with t(1;19)(q23;p13.3); TCF3-PBX1
T lymphoblastic leukemia
Immunophenotyping (Table 19.2)
Table 19.2
Immunophenotypic subgroups of childhood ALL
Antigen expression (% of cases positive) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Subtype | CD19 | CD22 | CD79a | CD10 | CD7 | CD5 | CD3 | cIgM | sIgM | sIg κ or λ | Prevalence (%) |
Early pre-B | 100 | >95a | >95 | 95 | 5 | 0 | 0 | 0 | 0 | 0 | 60–65 |
Pre-B | 100 | 100a | 100 | >95 | 0 | <2 | 0 | 100b | 0 | 0 | 20–25 |
B | 100 | 100a | 100 | 50 | 0 | 0 | 0 | >95 | >95 | >95 | 2–3 |
T | <5 | 0 | 30 | 45 | 100 | 95 | 100a | 0 | 0 | 0 | 15–18 |
B-Lineage ALL
B-lineage ALL is characterized by the expression of the B-cell markers CD19, CD22, and CD79α [76]. The cells lack expression of cytoplasmic (or surface) CD3 and of myeloperoxidase (by cytochemistry and antibody staining). Leukemic cells express various markers of differentiation that allow the distinction of at least three main subtypes: early pre-B ALL, pre-B ALL, and B-cell ALL.
Early pre-B ALL is characterized by the lack immunoglobulin synthesis. Thus, surface immunoglobulins are absent, and IgM heavy chains are not detectable in the cytoplasm. Expression of CD10 and terminal deoxynucleotidyl transferase (TdT) is found in at least 90 % of cases, while CD34 is expressed in more than 75 % of cases [76]. CD20 can be detected in approximately 50 % of cases [77]; although at diagnosis only a proportion of cells might be CD20 positive, its expression increases during treatment [78]. Early pre-B ALL cases with rearrangement of the MLL gene have a distinguishing immunophenotypic profile, including expression of chondroitin protoglycan sulfate, CD15, and/or CD65, and lack of CD10 [27, 79]. Weak or undetectable CD45 expression is common in cases with hyperdiploidy (>50 chromosomes) [80].
Pre-B ALL is defined by the presence of cytoplasmic IgM heavy chains with no detectable surface immunoglobulins and represents approximately 20–25 % of cases of B-lineage ALL [76]. In rare cases, IgM heavy chains without κ or λ light chains are also detectable on the cell surface; such cases have been designated transitional pre-B ALL [81], but this nomenclature is not widely used. Like early pre-B ALL, pre-B ALL cells usually express CD10 and TdT, with about two-thirds expressing CD34 [76]. ZAP-70 expression is more prevalent in cases with a pre-B phenotype [82, 83]. The TCF3-PBX1 gene fusion is typically found in cases with pre-B ALL phenotypes and characterizes 20–25 % of pre-B ALL cases [76].
B-cell ALL is defined by the expression of complete surface immunoglobulins. This phenotype occurs in 2–4 % of childhood ALL cases. Cells are generally recognizable by morphology owing to their FAB L3 appearance. CD20 and CD10 are often expressed and CD34 is negative [76]. B-cell ALL and the leukemic phase of Burkitt’s lymphoma are often indistinguishable. A characteristic of B-cell ALL is the presence of c-MYC gene rearrangements caused by the translocation of this gene on chromosome 8 with one of the chromosomes containing an immunoglobulin gene, such as t(8;14)(q24;q32), t(2;8)(p12;q24), and t(8;22)(q24;q11) [84]. A more uncommon subtype of B-cell ALL is characterized by blast cells with L1 or L2 morphology, expression of TdT and/or CD34, and lack of c-MYC rearrangements [76].
T-Lineage ALL
The markers most consistently expressed in T-lineage ALL cells and required for its diagnosis are CD7 and CD3. Expression of the latter is confined to the cytoplasm in approximately two-thirds of cases [85]; approximately half of these cases also express cytoplasmic TCR proteins (TCRβ, TCRα, or both). In the remaining third of T-ALL cases, CD3 is expressed on the cell surface together with TCR proteins, TCRαβ, or, less commonly, TCRγδ [86]. Other markers usually expressed in T-ALL include CD2, CD5, and TdT; CD1a is detected in approximately half of the cases and two-thirds express CD4 and/or CD8 [76]. Approximately 40 % of cases are CD10+ and/or CD21+, and CD79α is weakly expressed in about one-third of cases [76].
Using the immunophenotypes that define sequential stages of normal T-cell development in the thymus, it is possible to distinguish various stages of differentiation among T-ALL cases such as early T-ALL (surface CD3−, CD4−, and CD8−), mid or common (surface CD3−, CD4+, CD8+, and CD1a+), and late (surface CD3+, CD1a−, and either CD4+ or CD8+). However, many cases have a cell marker profile that does not totally fit poorly with this schema and this classification has not proven to be prognostically important in pediatric T-ALL [87, 88]. We used the gene expression profile characteristic of normal thymic T-cell precursor (ETP) cells, a population of recent immigrants from the bone marrow to the thymus which retains multi-lineage differentiation potential [89, 90], to search for T-ALL cases representative of this stage of T-cell differentiation. We found that leukemias with this gene expression profile (approximately 13 % of all T-ALL cases) lacked CD1a and CD8 expression, had weak or absent CD5 expression, and expressed at least one stem cell or myeloid-associated antigen (e.g., CD34, CD117, CD13, CD33, and CD11b) [91]. Importantly, ETP-ALL had an extremely poor response to chemotherapy [91].
Cytogenetics and Molecular Pathology (Table 19.3)
Table 19.3
Main genetic abnormalities in childhood B-lineage ALL and their relation to prognosis with contemporary therapy
Karyotypic abnormality | Molecular abnormality | Frequency | Prognostic impact |
|---|---|---|---|
Hyperdiploidy 51–65 chromosomes | 25% | Favorable | |
t(12;21)(p13;q22) | ETV6-RUNX1 | 22% | Favorable |
IKZF1 deletions or mutations | 15% | Unfavorable | |
t(4;11)(q21;q23) | MLL-AFF1 | 8% | Unfavorable in infants |
t(11;19)(q23;q13.3) | MLL-ELL | ||
t(X;14)(p22;q32) or t(Y;14)(p11;q32) | IGH@-CRLF2 | 5% | Unfavorable |
del(X)(p22.33p22.33) or del(Y)(p11.32p11.32) | CRLF2 deletions | ||
t (1;19)(q23;p13.3) | TCF3-PBX1 | 5% | Favorable |
t(9;22)(q34;q11) | BCR-ABL1 | 3% | Unfavorable |
Hypodiploidy <45 chromosomes | 1% | Unfavorable |
ALL with BCR-ABL1 Rearrangements
The BCR-ABL1 fusion gene results from the t(9;22)(q34;q11), which brings about the 5′ portion of BCR and the 3′ portion of ABL1, with N-terminal sequences of ABL1 being replaced by BCR sequences [92]. In ALL, breaks occur most often in the minor breakpoint cluster regions, resulting in a BCR-ABL1 fusion protein of 185–190 kD with a constitutively active ABL tyrosine kinase, which subverts normal signaling pathways and has profound effects on cell function [93]. Indeed, expression of the corresponding p185–190 DNA in transgenic mice causes leukemia (myeloid or lymphoid) 10–58 days after birth [94]. Moreover, mouse bone marrow cells transduced with retroviral vectors encoding p185–190 BCR-ABL1 induce B-cell leukemias when transplanted into lethally irradiated mice; the disease is significantly more aggressive if the Arf tumor suppressor is inactivated [95]. Genome-wide analysis of BCR-ABL1 ALL samples revealed deletions of IKZF1 in 84 % of cases; while these deletions were not found in chronic-phase chronic myelogenous leukemia (CML) samples, they were found in 70 % of CML in lymphoid blast crisis [34].
In addition to B-lineage ALL, ABL1 gene fusions can also occur in T-cell ALL and include NUP214-ABL1 (the most frequent, found in up to 6 % of cases), EML1-ABL1, BCR-ABL1, and ETV6-ABL1 [96].
ALL with MLL Gene Rearrangements
Structural alterations involving band q23 of chromosome 11 are the most frequent cytogenetic abnormalities in infant ALL, but they also occur in older patients [97]. The most common 11q23 abnormality in ALL is the t(4;11), which produces a gene fusion containing the N-terminal portion of the MLL gene (for mixed-lineage leukemia; also known as HRX, ALL-1, and TRX1) linked to the C-terminal portion of the AFF1 (alias AF-4) gene [98]. Many other translocations of 11q23 can occur and MLL has been shown to associate with more than 50 other genes. The MLL gene encodes a DNA-binding protein that regulates the expression of many other genes. Thus, cells in MLL-rearranged ALL have a characteristic gene expression profile, which differs from that of other ALL subtypes and also from that of other cases of infant leukemia [27, 99]. Inappropriately overexpressed genes include several genes normally expressed in non-lymphoid hematopoietic cells, such as FLT3, LMO2, and HOX genes (e.g., HOXA9, HOXA5, HOXA4, and HOXC6) [27]. The antiapoptotic gene MCL1 is also overexpressed [100]. The function of MLL is essential for embryonic development and hematopoiesis; MLL fusion proteins are sufficient to transform hematopoietic cells into leukemia-initiating cells [101]. Genome-wide SNP array analysis showed that gene copy number abnormalities are lacking in MLL-rearranged ALL, corroborating the notion that MLL abnormalities are a “first-hit” sufficient to drive the onset of leukemia [33, 102]. Recent studies showed significant promoter CpG island hypermethylation in MLL ALL compared to other ALL subtypes [103–105], with distinct methylation patterns between MLL-AFF1 and MLL-ENL or MLL-MLLT3 cases [104]. Treatment with the DNA methyltransferase inhibitor decitabine was cytotoxic to MLL-rearranged cells while causing re-expression of some of the gene silenced by methylation [105].
ALL with ETV6-RUNX1 Rearrangements
The ETV6-RUNX1 gene fusion is found in approximately 25 % of B-lineage ALL cases [106]. It is caused by the t(12;21)(p13;q22) translocation which juxtaposes the 5′ portion of the ETV6 (TEL) gene and the nearly complete RUNX1 (AML1) gene [107]. This translocation is usually cryptic and the abnormality can be detected only by fluorescence in situ hybridization (FISH) or reverse-transcriptase polymerase chain reaction (RT-PCR) [107]. The non-translocated ETV6 allele is frequently deleted.
The widely expressed ETV6 gene belongs to the Ets family of transcription factors and RUNX1 encodes a transcription factor that binds DNA as a heterodimer with core binding factor (CBF) β; both ETV6 and RUNX1 are essential for normal hematopoietic development [108, 109]. It appears that ETV6-RUNX1 requires cooperating genetic alterations to cause leukemia. Transduction of murine bone marrow cells with ETV6-RUNX1 was associated with the occurrence of leukemia resembling ALL in a minority of transplanted mice, but the frequency of leukemia development was increased by inactivation of Arf [110]. Mice in which ETV6-RUNX1 was driven from the endogenous promoter were rendered prone to malignancy after chemical mutagenesis when expressed in hematopoietic stem cells, but not in early lymphoid progenitors [111]. ETV6-RUNX1 increased the number of stem cells while predominantly maintaining them in cell cycle quiescence [111]. Expression of ETV6-RUNX1 alone in human cord blood progenitor cells led to the expansion of a candidate preleukemic population that had a growth advantage in the presence of TGF-beta [46, 112]. The ETV6-RUNX1 protein binds to Smad3, a TGF-beta signaling target, and results in a lower sensitivity to TGF-beta-mediated inhibition of proliferation [112]. In experimental cellular models, expression of ETV6-RUNX1 resulted in downregulation of MAD2L1 mRNA and a tetraploid karyotype, in line with findings of near-triploidy, near-tetraploidy, and trisomy 21 as secondary changes in ETV6-RUNX1 ALL [113].
ALL with TCF3 Rearrangements
Approximately 20–25 % of pre-B ALL cases have the t(1;19) (q23;p13) translocation abnormality which juxtaposes the TCF3 (E2A) gene on chromosome 19 and the PBX1 gene on chromosome 1 [114]. The resulting TCF3-PBX1 fusion protein contains the transcriptional activation domains of TCF3 linked to the DNA-binding domain of PBX1, thereby activating inappropriately the transcription of genes normally regulated by PBX1 [115]. Expression of the TCF3-PBX1 in mice leads to the development of lymphomas and myeloid leukemias [116].
Hyperdiploid ALL
In over a third of ALL cases, leukemic cells are hyperdiploid. Hyperdiploid cases with a chromosome number of 51–65 represent a distinct biologic subset of B-lineage ALL with an excellent prognosis [119–121]. Leukemic lymphoblasts with this karyotype have a marked propensity to undergo apoptosis [122], and accumulate greater quantities of methotrexate and its active polyglutamate metabolites [123]. A recent study investigated 74 cases of hyperdiploid ALL by SNP analysis and revealed a complex picture, with chromosomal abnormalities being more frequent than those detected by conventional karyotyping [124]. The same group investigated the genetic changes occurring during relapse using 11 paired diagnostic and relapse samples [125]. Structural changes, mainly cryptic hemizygous deletions, were significantly more common at relapse and deletions involving the IKZF1, PAX5, CDKN2A/B, or AK3 genes were recurrent.
Genetic Subtypes of T-cell ALL
Genes that are dysregulated in T-cell ALL include SCL (TAL-1), LMO1 (TTG-1), LMO2 (TTG-2), and HOX11 [24, 98]. In a small fraction of cases, the SCL gene (on chromosome 1p32) is inserted into the TCRδ locus on chromosome 14q11, but in a higher proportion of cases (approximately 25 %), there is an internal deletion in the 5′ untranslated region of SCL, which juxtaposes a locus called SIL with the SCL coding region, resulting in the expression of a fused SIL-SCL transcript, which leads to an inappropriate expression of the SCL protein [126, 127]. SCL encodes a helix-loop-helix transcription factor that is essential for the development of hematopoietic cells. LMO1 is inserted into the TCRA/D locus in the t(11;14)(p15;q11), while LMO2 is inserted into this locus in the t(11;14)(p13;q11) [128]. Activation of LMO2 by retroviral insertion in its proximity is involved in the development of T-cell ALL in patients with X-linked severe combined immunodeficiency who received hematopoietic stem cells transduced with the IL2GR gene [129, 130]. Lmo2 induced self-renewal of committed T cells in the mice more than 8 months before the development of overt T-ALL [131]. These self-renewing cells retained the capacity for T-cell differentiation but expressed several genes typical of hematopoietic stem cells, suggesting that Lmo2 might reactivate an HSC-specific transcriptional program. Forced expression of one such gene, Hhex, was sufficient to initiate self-renewal of thymocytes in vivo [131]. An additional alteration found in T-cell ALL is the deletion from chromosome 9p21of the INK4a and INK4b genes, which encode the p16INK4a and p15INK4b, inhibitors of the Cdk4 cyclin d-dependent kinase [132]. This locus, which is deleted in more than 50 % of cases, also encodes another important regulator of cell cycle and apoptosis, p19ARF [133].
A study reported inactivating mutations and deletions in the X-linked plant homeodomain finger 6 (PHF6) gene in 16 % of pediatric T-ALL cases, associated with aberrant expression of the homeobox transcription factor oncogenes TLX1 and TLX3 [134]. Alterations of PTEN, PI3K, or AKT were identified in approximately half of T-ALL cases [135]. LEF1 deletions or sequence alterations were also described in 16 % of T-ALL cases, which showed increased expression of MYC and MYC target, activating NOTCH1 mutations, biallelic INK4a/ARF deletions, and PTEN loss-of-function mutations or activating mutations of PI3K or AKT genes, and differentiation arrest at cortical stage of thymocyte development [136].
Levels of HOX11, TAL1, and LYL1 mRNA expression have been used to recognize distinct subtypes of T-ALL [31]. Overexpression of LMO1 or LMO2 was observed in most samples also overexpressing TAL1, and high levels of LMO2, but not LMO1, were found in cases with high LYL1 expression [31]. Activating mutations of NOTCH1 are frequently found in T-ALL and have been implicated in its pathogenesis [23], while expression of the chemokine receptor CCR7 has been shown to play an important role in CNS infiltration in murine models of T-ALL [137]. A study identified two T-ALL subtypes that lacked rearrangements of known oncogenes, one with ectopic NKX2-1 or NKX2-2 expression and the other with high expression of the MEF2C transcription factor [138].
Whole genome sequencing of tumor and normal DNA from 12 children with ETP ALL was recently performed; the frequency of the identified non-silent somatic mutations was determined in a separate cohort of 52 ETP and 42 non-ETP T-ALL cases [39]. A high frequency of inactivating mutations potentially disrupting lymphoid development and involving EP300, ETV6, GATA2, GATA3, IKZF1, and RUNX1 was identified in ETP ALL. In parallel, activating mutations in genes regulating cytokine receptor and RAS signaling (NRAS, KRAS, FLT3, IL7R, JAK3, JAK1, and BRAF) were demonstrated. Overall, the mutational spectrum in ETP ALL recalls that observed in acute myeloid leukemia.
Prognostic Factors
Clinical Presenting Features
Figure 19.1 summarizes the clinical and biologic features of children and adolescents with ALL enrolled in the St. Jude Children’s Research Hospital Total studies 11–13B and 15 [17, 139]. Historically, boys had worse outcome than girls [140–146], but in effective contemporary treatment protocols, there is no gender difference in treatment outcome [17, 147, 148]. Likewise, in many studies, patients of African–American and Hispanic origin had a significantly worse outcome than Caucasians [149, 150], but this was not the case in studies performed at St. Jude Children’s Research Hospital, where all patients have equal access to effective contemporary treatment, regardless of their family’s ability to pay [19, 151]. A recent study of genome-wide germline SNP genotypes showed that genomic variation that co-segregated with Native American ancestry was associated with a higher risk of relapse, but this association was abrogated by the addition of an extra phase of chemotherapy [152].
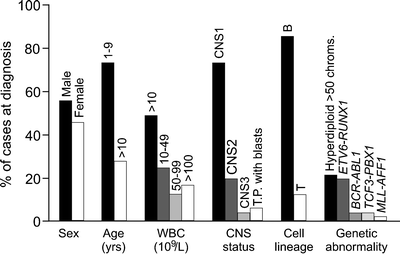
Age and leukocyte count at diagnosis provide prognostic information in B-lineage ALL but not in T-lineage ALL [144, 153, 154]. Thus, patients with B-lineage ALL are classified to be at a lower risk of relapse if age is between 1 and 9 years and leukocyte count <50 × 109/L. However, the reliability of this classification is somewhat limited as approximately 20 % of such patients may subsequently relapse and patients at a very high risk of relapse cannot be reliably identified. A recent study demonstrated that the adolescents (age 15–18 years) with ALL, a group that historically had a worse prognosis, can attain cure rates similar to those of younger patients with contemporary chemotherapy [155]. Infants (<1 year of age) with MLL-rearranged ALL, especially those under 6 months of age, have a dismal prognosis [140, 156]. Patients with detectable ALL blasts in the CSF (even from traumatic lumbar puncture) generally have an increased risk of CNS and hematological relapse and inferior overall outcome [72, 157–159].
Recent risk classification criteria adopted by different group of investigators are summarized in Fig. 19.2.

Fig. 19.2
Cellular and Genetic Features
Morphologic differences in leukemic cells (i.e., L1 versus L2 FAB classification) have no prognostic relevance. Among immunophenotypic features, patients with T-lineage ALL generally have a lower rate of event-free survival than those with B-lineage ALL [17], and/or a higher rate of induction failure, early relapse, and isolated CNS relapse [160]. Other immunophenotypic features, such as expression of myeloid-associated antigens, have no prognostic significance [161]. Among patients with T-ALL, it is important to identify those with ETP-ALL, as patients with this form of leukemia have a dismal outcome even with contemporary therapy and require more intensive and innovative treatment approaches [91].
Some genetic features of ALL are known to be associated with poor response to therapy. It is well known that BCR-ABL1 ALL carries a poor prognosis with standard chemotherapy; age over 10 years, a high leukocyte count, and poor early response are adverse prognostic features in this ALL subtype [162, 163]. However, intensive combination chemotherapy together with imatinib yielded an excellent 3-year event-free survival exceeding 80 % [164]. den Boer et al. [32] described a high-risk subset of ALL with a gene expression profile similar to that of BCR-ABL1-positive cases. The majority of cases with BCR-ABL1-like disease also had deletions in genes involved in B-cell development such as PAX5 and IKZF1. Mullighan et al. [165] independently showed that IKZF1 deletions and mutations were associated with a BCR-ABL1-like gene expression profile, poor early treatment response with high levels of minimal residual disease (MRD) after induction therapy, and high risk of relapse. Among patients with B-lineage ALL and high-risk clinical features, those with JAK mutations or CRLF2 rearrangements reportedly have a significantly poorer outcome [166]. The prognostic impact of MLL gene rearrangement differs by age group, with infants having the worst outcome [97, 156, 167, 168]. Hypodiploidy (<45 chromosomes) occurs in <2 % of ALL cases and is associated with a poor outcome [169, 170]. Finally, patients with TCF3-HLF ALL often present with hypercalcemia and coagulopathy, and have poor prognosis [171].
Other genetic features are associated with favorable prognosis. In most studies, patients with ETV6-RUNX1 ALL had an excellent prognosis [17, 121, 172–175]. In agreement with this clinical response is the increased in vitro sensitivity to several antileukemic drugs observed in some studies [176, 177]. Nevertheless, ETV6-RUNX1 ALL cells have a reduced capacity to accumulate methotrexate polyglutamates overall, suggesting that an increased dose or prolonged infusion of methotrexate might be beneficial [178]. Another genetic subtype of ALL with favorable prognosis is hyperdiploid ALL (>50 chromosomes) [179–182]. Among chromosomes that are overrepresented, only the trisomies of chromosome 4, 10, and 17 were associated with a favorable prognosis in some studies [121]. In contrast to the overall favorable prognosis of cases with 51–65 chromosomes, ALL cases with near-triploidy (69–81 chromosomes) have a response to therapy similar to that of non-hyperdiploid ALL [183]. Cases with near-tetraploidy (>80 chromosomes) have a high frequency of T-cell immunophenotype, are associated with ETV6-RUNX1 when they occur in B-lineage, and have a relatively favorable outcome [184]. With contemporary treatment protocols, patients with TCF3-PBX1 ALL have excellent response to initial therapy [17] and a favorable overall outcome, although they still have increased risk of leukemia relapse in the CNS [185].
Among cases of T-ALL, those bearing the gene expression profile of ETP have a dismal outcome [91]. High expression of HOX11L2 has been associated with poor outcome in some studies [31, 186], but not in others [187]. In most studies, NOTCH1 mutations in T-cell ALL were either associated with a favorable prognosis or had no prognostic impact [88, 188–191].
Pharmacogenetic Variables
Approximately 1 in 300 patients has an inherited homozygous deficiency of thiopurine S-methyltransferase, which catalyzes the inactivation of mercaptopurine, and is therefore extremely sensitive to this drug [192]. Patients who are heterozygous for this mutation (approximately 10 %) have intermediate levels of enzyme activity [192]. Both homozygous and heterozygous deficiencies are associated with a better event-free survival, likely due to higher dose intensity of mercaptopurine, but they also carry the increased risk of acute hematopoietic toxicities, and a higher rate of irradiation-induced brain tumors and therapy-related acute myeloid leukemia [192–195]. Therefore, it is important to adjust mercaptopurine dosages carefully in these patients [192].
The null genotype of glutathione S-transferase (GST)M1 or GSTT1 and the GSTP1 Val 105 /Val 105 genes is also associated with increased treatment-related toxicity [196, 197], and a lower risk of relapse [198], presumably because of the reduced detoxification of chemotherapy. Sixteen polymorphisms in genes involved in drug metabolism, including the GSTM1 and thymidylate synthetase (TYMS), were associated with treatment outcome in one study [199]. In another study, polymorphisms in genes involved in folate metabolism, including methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR), affected methotrexate sensitivity in vitro [200]. Davies et al. [201] found that the G allele of a common polymorphism in chemokine receptor 5 (CCR5) gene was associated with a better leukemia clearance during remission induction therapy than the A allele; the molecular mechanisms underlying this association are unclear. Yang et al. [202] identified 102 SNPs associated with MRD, including 21 in genes associated with antileukemic drug disposition and 5 in the interleukin (IL) 15 gene. The mechanisms linking alterations in IL-15 and poor response to chemotherapy are also unclear, but this finding is in line with a report pointing to an association between IL-15 overexpression and increased CNS involvement at diagnosis as well as CNS relapse [203]. Significantly higher concentrations of methyl mercaptopurine nucleotides were found in patients with a nonfunctional allele of the inosine triphosphate pyrophosphatase gene, which were associated with a higher probability of severe febrile neutropenia [204]. SNPs in the organic anion transporter polypeptide SLCO1B1 were associated with methotrexate’s pharmacokinetics and gastrointestinal toxicity [205].
Minimal Residual Disease
The persistence of lymphoblasts in bone marrow or peripheral blood samples taken from patients with ALL during remission induction therapy indicates a suboptimal response and is associated with a significantly increased risk of relapse [140, 206]. Indeed, risk-directed therapy based on classification schemas including early treatment response appears to improve outcome [207]. However, traditional morphological assessment of treatment response can be imprecise and lack sensitivity because the morphology of ALL cells is very similar to that of immature lymphoid precursors and activated lymphocytes [208]. The advent of assays to detect leukemic cells undetectable by morphology, i.e., MRD, has dramatically improved the precision and sensitivity of treatment response evaluations. MRD in ALL can be studied by flow cytometric detection of aberrant immunophenotypes (expressed by leukemic cells in approximately 95 % of patients), polymerase chain reaction (PCR) amplification of immunoglobulin (IG) and TCR genes (clonally rearranged in approximately 90 % of patients), or PCR amplification of fusion transcripts (detectable in approximately 40 % of patients). These methods allow the detection of one leukemic cell among 10,000–100,000 normal bone marrow or peripheral blood cells [208].
Flow cytometry was used at St. Jude Children’s Research Hospital to prospectively monitor MRD in children with newly diagnosed ALL enrolled in the Total XIII study [209–211]. MRD (i.e., ≥0.01 %) at any of the point during treatment predicted a higher risk of relapse, and MRD ≥1 % at the end of remission induction therapy was associated with a particularly poor outcome. By contrast, MRD <0.01 % on day 19 of remission induction therapy was strongly associated with subsequent continuous complete remission: the 3-year cumulative incidence of relapse for patients with this finding was only 1.9 %. The Children’s Oncology Group also used flow cytometry to monitor MRD in bone marrow collected on day 29 (end of remission induction therapy) in children with B-lineage ALL [212]. MRD ≥0.01 % was associated with shorter event-free survival in all risk groups. The Italian cooperative group AIEOP studied bone marrow samples on day 15, after 14 days of steroids, and one dose of intrathecal methotrexate, vincristine, daunorubicin, and asparaginase, and identified three risk groups: standard (<0.1 % MRD; 42 %), intermediate (0.1 %- <10 % MRD; 47 %), and high (≥10 %; 11 %) with a 5-year cumulative incidence of relapse of 7.5 %, 17.5 %, and 47.2 %, respectively [213].
The European Organization for Research and Treatment of Cancer (EORTC) protocol studied MRD by PCR amplification of IG/TCR genes in children with ALL and found that levels of MRD during the first 6 months of therapy significantly predicted the risk of relapse: patients with ≥1 % MRD after remission induction therapy, or ≥0.1 % or higher at subsequent time points, had a particularly high risk of relapse [214]. The International BFM Study Group used combined information from MRD measurements performed on day 33 and day 78 to identify three groups of patients with a different risk of relapse: a standard-risk group (43 % of patients) with a 3-year relapse rate of 2 %; a high-risk group (15 %) with a relapse rate of 75 %; and an intermediate-risk group (43 %) with a 3-year relapse rate of 23 % [215], and a 10-year event-free survival of 93 % for standard-, 16 % for the high-, and 74 % for the intermediate-risk group [216]. Two or more targets that allow PCR analysis with a 1 in 10,000 sensitivity or better were identified in 71 % of patients, while an additional 20 % had only one such target [216]. Among patients with B-lineage ALL, those considered to have standard-risk by MRD criteria (42 %) had a 5-year event-free survival of 92.3 %; the event-free survival rates were 50.1 % for the high-risk patients (6 %) and 77.6 % for those with intermediate-risk ALL (52 %) [217]. In a study of patients with B-lineage ALL by the Dana–Farber Cancer Institute ALL Consortium, the 5-year risk of relapse was 5 % for the 62 % of the patients with no detectable MRD at the end of remission induction therapy and 44 % for those with detectable MRD [218]. Finally, a recent study at St. Jude Children’s Research Hospital demonstrated that even very low levels of MRD (i.e., between 0.001 % and 0.01 %) as detected by PCR amplification of IG/TCR genes at the end of remission induction therapy are associated with an increased risk of relapse [219].
Levels of MRD during induction therapy are associated with genetic features of the disease. Patients with BCR-ABL1 ALL, IKZF1 mutations, and ETP-ALL have a high prevalence of MRD, while those with TCF3-PBX1 and ETV6-RUNX1 most commonly have lower levels or undetectable MRD [91, 165, 220]. Despite these associations, MRD was an independent prognostic indicator in all the noted studies. In addition, MRD was significantly associated with outcome in studies limited to specific subtypes of ALL. Thus, in a recent study, all infants with levels of MRD (detected by PCR of IG/TCR and MLL genes) ≥0.01 % at the end of consolidation relapsed, while only 13 % of patients with MRD <0.01 % at both the end of induction and end of consolidation relapsed; the remaining patients had a relapse rate of 31 % [221]. Of note, infant MLL-AFF1 ALL also has a high prevalence of immature, nonproductive, and/or oligoclonal antigen–receptor gene rearrangements [167, 168], and it may be difficult to identify suitable IG/TCR rearrangements for monitoring MRD. In these cases, flow cytometry and PCR amplification of the MLL-AFF1 fusion transcripts can often be used instead.
Because of their strong predictive power, measurements of early treatment response according to MRD have been incorporated into clinical trials to refine risk assignment [17, 216]. Thus, MRD studies by flow cytometry or PCR IG/TCR were performed in all 498 evaluable patients enrolled in the St. Jude Total XV study, where MRD testing was a key part of the risk classification schema [17]. This study yielded 5-year event-free and overall survival probabilities of 85.6 % and 93.5 %, respectively, suggesting that the use of MRD contributed to improved outcome. However, despite treatment intensification for MRD-positive patients, high MRD (≥ 1 %) at the end of induction remained an independent predictor of poor event-free survival.
Treatment
Treatment for patients with B-lineage (early pre-B and pre-B) and T-lineage ALL is generally constituted by three main phases: remission induction, intensification (consolidation) therapy, and continuation therapy. The more successful contemporary clinical trials (Table 19.4) have resulted in 5-year event-free survival rates ranging from 75 % to 87 % [17, 139, 143–145, 148, 222–239]. In patients with mature B-cell ALL, regimens of intensive chemotherapy are typically shorter and based primarily on cyclophosphamide, methotrexate, and cytarabine, resulting in cure rates of 74–87 % [240–242]. All patients require treatment for subclinical leukemia of the CNS, which should be initiated early in the form of intrathecal therapy.
Table 19.4
Results of selected clinical trials in childhood ALL
Study | Time period | No. of patients | % 5-year event-free survival (± SE) | References |
|---|---|---|---|---|
DFCI-95–01 | 1996–2000 | 491 | 82 ± 2 | [226] |
BFM 95 | 1995–2000 | 2169 | 78.6 ± 0.9a | [228] |
CCG-1961 | 1996–2002 | 2078 | 71.3 ± 1.6b | [227] |
COALL-97 | 1997–2003 | 667 | 77 ± 2 | [232] |
NOPHO ALL-2000 | 2000–2007 | 1023 | 79.4 ± 1.5 | [235] |
SJCRH XV | 2000–2007 | 498 | 85.6 ± 2.9 | [17] |
Remission Induction Therapy
Remission induction therapy typically includes a glucocorticoid (prednisone, prednisolone, or dexamethasone), vincristine, and asparaginase; one or more other drugs, such as an anthracycline and cyclophosphamide, may be added. In general, the clearance of leukemic cells by remission induction therapy is proportional to its intensity, but responses depend on the biological features of leukemic cells and intensification may increase early morbidity and mortality [243, 244]. Thus, intensified induction treatment might not be required for patients lacking unfavorable prognostic features and for those showing a good response to the initial phases of induction therapy, particularly if they receive post-induction intensification therapy [141, 142].
Among glucocorticoids, dexamethasone may be more effective than prednisone or prednisolone [231, 245]. Its use yielded an improved outcome in two randomized trials [246, 247] but resulted in excessive life-threatening infections and septic deaths in another study [244]. A small randomized study showed that an increased dose of prednisone produced results comparable to those achieved with dexamethasone in the context of other intensive treatment, suggesting that dose rather than type of glucocorticoid influences treatment outcome [248]. Pharmacologic advantages of dexamethasone include its longer half-life and increased penetration into CSF [249]. Notably, a recently reported study of the Children’s Cancer Group (CCG) failed to demonstrate any meaningful differences in long-term cognitive functioning for patients with childhood ALL randomized to receive dexamethasone or prednisone [250].
The key endpoint to predict the antileukemic activity of l-asparaginase is the extent of asparagine depletion [251]. Asparaginase administration has also been associated with toxicities, including thrombosis, hypersensitivity reaction, and pancreatitis. Risk factors for thrombosis include concomitant steroid use, presence of central venous lines, and thrombophilic abnormalities [252]. Currently available forms of asparaginase for first-line use are derived from Escherichia coli, one as polyethylene glycol (PEG) compound; asparaginase derived from Erwinia chrysanthemi is typically used in patients who have hypersensitivity reactions to E. coli product [253, 254]. Each type has a different half-life, pharmacokinetics properties, and immunogenicity [255]. Asparaginase has typically been administered intramuscularly in the USA. A recently reported Dana–Farber Cancer Institute ALL Consortium Protocol 05-01study evaluated the feasibility of administering PEG-asparaginase intravenously (2,500 IU/m2 over 1 h) during remission induction therapy [256]. Serum asparaginase activity >0.1 IU/mL was detected in 95 %, 88 %, and 7 % of patients at 11, 18, and 25 days after dosing, respectively; toxicities included allergy (1.5 %), venous thrombosis (2 %), and pancreatitis (4.6 %), indicating that intravenous administration is effective and tolerable. Because of lower immunogenicity, less frequent administration due to long half-life, and feasibility to administer intravenously [147, 257], PEG-asparaginase has become the first-line treatment in the USA, and is also increasingly used worldwide. Although l-asparaginase is typically included in remission induction therapy, clinical trials limiting its use to the post-induction phase period have also yielded excellent overall outcome with low morbidity, particularly in regard to thrombotic complications [142, 147].
With contemporary remission induction therapy, complete remission, defined as the absence of leukemic cells detectable morphologically and restoration of normal hematopoiesis, can be achieved in 96–99 % of patients [17, 139, 143–145, 148, 222–228, 231–233, 235, 237, 238]. Improvements in supportive care have reduced the early death rate to less than 2 % [258].
Intensification (Consolidation)
After achieving remission, patients usually receive intensification (alias consolidation) therapy. This may be the same drug regimen used for remission induction therapy or a combination of drugs not used in the previous phase. Some intensification regimens appear to be particularly effective for some leukemia subtypes. Thus, patients with T-ALL benefited from very high dose of methotrexate (5 g/m2) in one trial [259], while those with ETV6-RUNX1 benefited from intensive post-remission treatment with glucocorticoids, vincristine, and asparaginase in other trials [17, 148, 260]. Conversely, other intensification regimens, such as the use of high-dose intravenous mercaptopurine or high-dose cytarabine, showed no advantage [223, 246, 261, 262]. A recent study showed that shortening the infusion time of high-dose methotrexate from 24 h to 4 h reduces its antileukemic effect, particularly for patients with T-ALL [263].
Delayed intensification (or reinduction) was first tested by the Berlin–Frankfurt–Münster consortium [259], and is now widely used because this treatment is beneficial to all patients. It is the repetition of remission induction therapy 3 months after the end of remission induction. In studies performed by the AIEOP group and the CCG, double delayed intensification improved patient outcome in patients with intermediate-risk ALL [264, 265]. In a CCG trial, augmented (rather than standard) post-induction intensification therapy with additional doses of vincristine and asparaginase (in lieu of 6-mercaptopurine during interim maintenance therapy) and increased dose of intravenous methotrexate without leucovorin rescue improved outcome in patients with high-risk ALL and slow response to initial induction therapy [266]. In a subsequent CCG study, there was no statistical difference in treatment outcome between patients with a rapid early response who were randomized to receive either one or two courses of post-induction intensification treatment [227, 267]. Additional studies are necessary to determine if prolonged post-induction intensification could benefit patients with slow early response.
Continuation Therapy
Prolonged continuation treatment, typically lasting 2–2.5 years, is required for all patients with ALL, except those with B-cell ALL. Shortening the duration of consolidation therapy resulted in inferior outcomes, although it is important to note that in one trial maintenance therapy for 6 months with all therapy stopping at 1 year resulted in nearly 60 % 5-year event-free survival, suggesting that such short therapy could be curative for more than a half of patients with ALL [268]. Continuous uninterrupted therapy appears to be more effective than high-dose pulse therapy with prolonged rest periods to recover from myelosuppression [147, 259, 266].
Methotrexate (administered weekly) and mercaptopurine (administered daily) are the standard components of continuation therapy. Tailoring of doses calculated to the limits of tolerance has been reported to improve outcome [269], but too strict adherence to such plan may result in dose interruption and reduction of the overall dose intensity [270]. Low systemic exposure to methotrexate and low-dose intensity of mercaptopurine have been associated with an inferior outcome [270, 271]; in a recent study, the degree of myelosuppression during maintenance was shown to be significantly associated with risk of relapse in adolescents [272]. In the case of methotrexate, decreased bioavailability and poor compliance associated with oral administration can be avoided by parenteral administration [273]. Mercaptopurine, however, is most effective when given orally; weekly intravenous administration at a higher dose was shown to be ineffective [147, 223, 246, 273]. In a recently reported study, patients with low-risk ALL (defined as those with age 1–9 years, leukocyte counts <50 × 109/L at diagnosis and <5 × 109/L on day 7 of therapy, no peripheral blasts and <25 % blasts in bone marrow on day 14, and <5 % blasts in the bone marrow on day 28) were randomized to receive either continuous 6-mercaptopurine and weekly methotrexate or intermittent 6-mercaptopurine (100 mg/m2/day for 10 days, with 11 days resting) with intermediate-dose methotrexate (200 mg/m2) every 3 weeks: the latter was less toxic and resulted in better event-free survival in boys but not in girls [274].
In some studies, thioguanine appeared to be superior to mercaptopurine in terms of antileukemic effect but was associated with a higher rate of toxicities, such as profound thrombocytopenia and hepatic veno-occlusive disease, when used daily at >40 mg/m2 [275–277]. In a recent meta-analysis of all randomized trials comparing thioguanine to mercaptopurine from the USA, UK, and Germany, the overall event-free survival was not significantly improved with thioguanine [278]. Intensive treatment with mercaptopurine and methotrexate has been correlated with the occurrence of second malignancies, especially in patients with thiopurine methyltransferase deficiency [279].
Studies have shown that adding intermittent pulses of vincristine and a glucocorticoid (prednisone or dexamethasone) to the methotrexate/mercaptopurine backbone was beneficial [141, 280]. However, prednisone and vincristine pulses failed to improve outcome in a CCG study that included reinduction treatment [264], and in a study of intermediate-risk patients treated on a BFM regimen [281]. Thus, it is not clear whether the pulse therapy is necessary in contemporary regimens including early intensification.
Prevention and Therapy of CNS Leukemia
Leukemic cells that infiltrate the leptomeninges are protected from systemic therapy by the blood–brain barrier [282, 283]. Therefore, cranial irradiation (12–24 Gy) and intrathecal methotrexate have been used since the 1970s as prophylactic measures to prevent CNS relapse [284]. However, cranial irradiation can cause serious side effects, prompting investigators to use a 12 Gy dose which appeared to provide adequate protection while being less harmful [259]. Complete omission of cranial irradiation as tested in early clinical trials had resulted in an acceptable rate of CNS relapse but produced low event-free survival and overall survival rates [143, 285]. A recent study at St. Jude Children’s Research Hospital tested the feasibility of total omission of prophylactic cranial irradiation [17]. The 5-year survival rate for the 498 patients enrolled was 93.5 % and the cumulative risk of an isolated CNS relapse rate was only 2.7 %, demonstrating that cranial irradiation can be avoided in the context of effective risk-adapted intrathecal and systemic chemotherapy. Importantly, patients with an isolated CNS relapse have a very high retrieval rate if they did not receive cranial irradiation as CNS prophylaxis [17, 286, 287]. In fact, all 11 patients with isolated CNS relapse in the St. Jude study remained in second remission after completion of salvage therapy. Dutch investigators independently confirmed that prophylactic cranial irradiation can be totally omitted in their recently completed ALL-9 study, which yielded a 5-year event-free survival of 81 % and a cumulative risk of isolated CNS relapse rate of 2.6 % [245]. Intensive systemic and intrathecal treatment, without cranial irradiation, appear to provide adequate CNS prophylaxis even in infants who have CNS leukemia at diagnosis [288–290]. Because infants are particularly vulnerable to CNS irradiation toxicities, most investigators now omit cranial irradiation in these cases.
Systemic treatment with high-dose methotrexate, intensive asparaginase, and dexamethasone as well as optimal intrathecal therapy is crucial for CNS leukemia control [73]. Triple intrathecal therapy with methotrexate, cytarabine, and hydrocortisone is more effective than intrathecal methotrexate alone in preventing CNS relapse [291]. In patients with detectable ALL blasts in the CSF (even because of traumatic lumbar puncture), intrathecal therapy should be intensified as this feature carries an increased risk of CNS relapse and inferior overall outcome [72, 158, 159].
Allogeneic Hematopoietic Stem Cell Transplantation
Patients with ALL that has features predictive of a very high risk of relapse are candidates to receive allogeneic HSCT after they achieve clinical remission. Features used as an indication for HSCT historically included poor response to initial therapy as shown by the presence of MRD ≥1 % at the end remission induction therapy, and an ALL subtype associated with a high risk of relapse such as BCR-ABL1 ALL and ETP-ALL [17, 91, 162, 163, 292, 293]. However, these features should be continuously reevaluated according to the results of treatment changes and the availability of new agents. In the cases of BCR-ABL1 ALL, for instance, the advent of tyrosine kinase inhibitors has drastically improved early treatment response [164], questioning the appropriateness of HSCT in first remission in these cases. Transplantation has not been shown to improve outcome of other subtypes of very high-risk leukemia, including infant cases and those with MLL rearrangement [97, 294, 295].
Treatment Sequelae
Cranial irradiation may cause second malignancies, as well as neuro-cognitive deficits and endocrine abnormalities, leading to obesity, short stature, precocious puberty, and osteoporosis [298–301]. Remission induction therapy that includes prednisone, vincristine, and asparaginase causes hyperglycemia in 10–20 % of patients; risk factors include adolescent age, obesity, family history of diabetes mellitus, and Down syndrome [302]. This induction regimen can also cause cerebral and/or peripheral vein thromboses in 3–5 % of the patients [303]. High cumulative doses of anthracyclines can result in severe cardiomyopathy, especially in younger patients [304, 305]. Intensive use of methotrexate and glucocorticoids has been associated with an increased risk of neurotoxicity and osteonecrosis [306–308].
Relapse
Leukemia may relapse during treatment or after cessation of therapy; in the latter case, relapse occurs usually within 2 years, but relapses occurring as late as 10 years after diagnosis have been reported [309]. The most common site of relapse is the bone marrow; relapse can also appear to be isolated to extramedullary sites, such as the CNS and testes, or involve both bone marrow and extramedullary sites. The so-called isolated extramedullary relapse, however, is often associated with the presence of submicroscopic leukemia in the bone marrow [310, 311].
Early studies indicated that morphologically and immunophenotypic shifts to AML could occur at relapse in some cases while cells maintained the karyotypic and molecular features of the original clone [312, 313]. Using increasingly sophisticated multiparameter flow cytometric methods, it is now possible to distinguish minor subpopulations of cells with an ambiguous immunophenotype at diagnosis which become prominent at relapse. For example, some cases of ETP-ALL that relapse might appear at first glance as AML, although the ETP immunophenotype is clearly revealed by a closer scrutiny [91]. Notably, a recent study found that in 8 of 22 cases of late-relapse T-ALL TCR gene rearrangement sequences had completely changed from diagnosis, and there was a different pattern of genomic abnormalities, suggesting a second T-ALL rather than a resurgence of the original clone [314]. Novel insights on the genetic evolution of ALL at relapse were revealed by a study of Mullighan et al. [315] who performed genome-wide DNA copy number analyses on matched diagnosis and relapse samples from 61 children with ALL and found that most relapse samples lacked some of the genetic abnormalities present at diagnosis. Backtracking studies revealed that cells corresponding to the relapse clone were often present as minor subpopulations at diagnosis. It was found that 18 % of relapse cases had mutations of CREBBP, which encodes the transcriptional coactivator and histone acetyltransferase CREB-binding protein, resulting in impaired histone acetylation and transcriptional regulation of CREBBP targets, including glucocorticoid responsive genes [316]. The mutations were also detected in subclones at diagnosis, suggesting that these events could contribute to drug resistance. Studies of the genetic features of ALL cells at the single cell level revealing a complex nonlinear, branching structure of the leukemic clone and sub-clones with distinctive genetic signatures have led Greaves to propose a “Darwinian” model of clonal evolution that includes a genetic heterogeneity of propagating or “stem” cells [317].
Bone marrow relapse, with or without extramedullary involvement, is associated with an unfavorable outcome and, somewhat counterintuitively, patients with isolated bone marrow relapse generally do worse than those with combined bone marrow and extramedullary relapse [318, 319]. Adverse risk factors for relapsed ALL include short initial remission and relapse on therapy, T-cell immunophenotype, BCR-ABL1 ALL, presence of circulating blast cells, or a high leukocyte count at relapse [320–325]. A review of patients with relapsed and refractory ALL previously treated at institutions of the Therapeutic Advances in Childhood Leukemia Consortium indicated complete remission rates of 83 ± 4 % for early first marrow relapse, 93 ± 3 % for late first marrow relapse, 44 ± 5 % for second marrow relapse, and 27 ± 6 % for third marrow relapse, and 5-year disease-free survival rates in CR2 and CR3 of 27 ± 4 % and 15 ± 7 %, respectively [326]. The presence of MRD at the end of second remission induction is a strong adverse prognostic indicator, especially in patients with a first relapse off-therapy [327–329]. Data from the Children’s Oncology Group also using flow cytometry have confirmed the importance of MRD as a prognostic factor in patients with relapsed ALL: 12-month event-free survival was 58 % for patients with MRD ≥ 0.01 % at the end of the first block of reinduction versus 80 % for those who were MRD negative [330]. Chemotherapy may be sufficient to induce prolonged second remissions in patients without high-risk features, but allogeneic HSCT is an option to be considered for the remaining patients, particularly those with high-risk features. The Acute Lymphoblastic Leukemia Relapse Berlin–Frankfurt–Münster (ALL-REZ BFM) Study Group reported the results of a prospective trial including 91 children with relapsed ALL receiving stem-cell transplantation. Probability of event-free survival posttransplant was significantly associated with MRD burden before transplant, and MRD was the only independent predictive parameter [297].
Future Directions
The current success of ALL therapy is the combined product of improvements in risk classification, chemotherapy, transplantation, and supportive care. Despite this remarkable progress, some leukemia subtypes still respond poorly to intensive therapy; for patients with these forms of ALL, substantial improvements can only come from the development of new agents and treatment modalities. New formulations of existing agents may improve efficacy and decrease toxicity [258, 331]. For example, nucleoside analogs, such as gemcitabine, clofarabine, and nelarabine, have shown some promise [331, 332]. Molecularly targeted therapy with imatinib mesylate and other ABL kinase inhibitors is now standard for patients with BCR-ABL1 ALL and has dramatically changed early treatment results [164]. Other available novel agents with promising preclinical results which have not yet been thoroughly explored clinically include inhibitors of FLT3 [333, 334], and a Bcl-2 homology domain 3 mimetic [335]. Inhibition of activated NOTCH1 in T-ALL with gamma-secretase inhibitors has produced disappointing responses and high toxicity, which may be attenuated by concomitant administration of glucocorticoids [336]. An inhibitor that targets the NOTCH transactivation complex has been developed, but clinical results are not yet available [337]. In addition to classical pharmacologic sanctuaries (e.g., the CNS) where leukemic cells can be protected from chemotherapy, the bone microenvironment itself can also protect ALL cells from chemotherapy [338, 339]. Thus, strategies designed to mobilize ALL cells should be explored.
Immunotherapies for ALL are becoming increasingly available. Rituximab (anti-CD20), gemtuzumab ozogamicin (anti-CD33), alemtuzumab (anti-CD52), and epratuzumab (anti-CD22) have already been included in some clinical trials [331, 340]; a recombinant immunotoxin consisting of the single-chain variable region of an anti-CD22 antibody fused to a 38-kDa fragment of Pseudomonas aeruginosa exotoxin A [341, 342] and a bispecific anti-CD3-anti-CD19 antibody derivative [343, 344] are also being tested. For patients receiving HSCT, donors can be selected to maximize natural killer cell alloreactivity, an approach that appears to reduce the relapse rate posttransplant [345, 346], and infusions of haploidentical NK cells are being evaluated [347, 348]. Chimeric receptors, composed of single-chain variable domain of murine antibodies and human signaling molecules, can redirect the specificity of autologous or allogeneic immune cells against ALL cells and may therefore enhance the effectiveness of T- and NK cell infusions [349–351].
The current prognostic classification of ALL could be further improved by deepening the understanding of oncogenetically important genetic lesions, a realistic objective in the face of early results using novel genome-wide screening techniques [39, 165, 352]. Such studies may also lead to the discovery of new molecular determinants of treatment response [220, 353–361]. This knowledge coupled with the increasingly sophisticated monitoring of treatment response allowed by MRD assays should result in improved risk classification and, ultimately, better cure rates with fewer toxicities.
References
1.
2.
Campana D, Janossy G. Proliferation of normal and malignant human immature lymphoid cells. Blood. 1988;71(5):1201–10.PubMed
3.
Hayhoe FG. Leukaemia. Research and clinical practice. London: J&A Churchill Ltd; 1960.
4.
Virchow R. Weisses Blut und Milztumoren. Med Ztg. 1847;16:9–15.
5.
Neumann E. Ueber Myelogene Leukämie. Berl klin Wschr. 1878;15:69–131.
6.
Ehrlich P. Histology of the blood, normal and pathologic. Nothnagel’s encyclopaedia of practical medicine (English edition). Philadelphia, PA: W.B. Saunders; 1905.
7.
Bright R. Observations on abdominal tumours and intumescence; illustrated by cases of disease of the spleen. Guy’s Hosp Rep. 1838;3:401.
8.
9.
Proceedings of the First Clinical ACTH Conference. Philadelphia: Blakiston Co.; 1950.
10.
Rosenthal MC, Saunders RH, Schwartz LI, Zannos L, Perez Santiago E, Dameshek W. The use of adrenocorticotropic hormone and cortisone in the treatment of leukemia and leukosarcoma. Blood. 1951;6(9):804–23.PubMed
11.
Hitchings GH, Elion GB. Layer on layer: the Bruce F. Cain memorial award lecture. Cancer Res. 1985;45(6):2415–20.PubMed
12.
13.
15.
16.
17.
18.
Ries LA, Melbert D, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2005, Bethesda, MD: National Cancer Institute; 2007. Based on November 2007 SEER data submission, posted to the SEER web site 2008 [serial online].
19.
Pui CH, Sandlund JT, Pei D, et al. Results of therapy for acute lymphoblastic leukemia in black and white children. J Am Med Assoc. 2003;290(15):2001–7.CrossRef
20.
Spector LG, Ross JA, Robison LL, Bhatia S. Epidemiology and etiology. In: Pui CH, editor. Childhood leukemias. 2nd ed. Cambridge: Cambridge University Press; 2006. p. 48–66.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
den Boer ML, van Slegtenhorst M, de Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34.CrossRef
33.
34.
35.
36.
37.
38.
39.
Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63.
40.
Couto E, Chen B, Hemminki K. Association of childhood acute lymphoblastic leukaemia with cancers in family members. Br J Cancer. 2005;93(11):1307–9.PubMedCrossRef
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


