Fig. 37.1
An enlarged tongue in a patient with AL amyloidosis
Fig. 37.2
Clinical presentation of classic vascular purpura
The Congo Red Stain
The only accepted diagnostic test to validate the diagnosis of amyloidosis is identification of amyloid deposits in biopsy specimens showing affinity for Congo red stain and demonstrating green birefringence under polarized light [5]. Use of the Congo red stain is not simple. Overfixation of biopsy specimens can result in poor staining, and trapping of the Congo red dye can result in false-positive results. The presence of fibrin and elastin in skin and fat tissues can bind Congo red and result in a false-positive interpretation. It has been suggested that phenolic Congo red may be superior [6]. Over the years, alternative methods to improve the specificity of Congo red have been attempted. Potassium permanganate digestion had previously been used to distinguish AA from other forms of amyloidosis. However, this has been abandoned for immunohistochemical, immunogold, and mass spectroscopic analysis of amyloid deposits. Congo red birefringence has been combined with immunocytochemical analysis to improve the specificity of the technique, although the reliability of this technique has recently been challenged [7, 8].
Biopsy Diagnosis of Amyloidosis
Amyloidosis is a widespread deposition disorder, and deposits throughout the vascular system are characteristic. As a consequence, deposits can be located via biopsy at virtually any site, which has increased the popularity of skin, fat, and gingival biopsies because of their accessibility. In clinical practice, biopsies are highly sensitive because they are generally directed to the clinically affected organ: the liver when hepatomegaly is present, the kidney when proteinuria in the nephrotic range is present, and so forth. Endomyocardial biopsy for cardiac amyloidosis is 100 % sensitive when sampling error is eliminated by obtaining at least four samples [9]. In one study of 36 renal biopsies, all specimens showed fibrillary deposits of amyloid with electron microscopy and stained positively with Congo red [10]. A typical electron micrograph illustrating amyloid fibrils is found in Fig. 37.3a. Amyloidosis can be diagnosed with fine-needle aspiration and has been used for liver biopsy diagnosis, reducing the risk of bleeding associated with core needle biopsies of liver tissue [11].
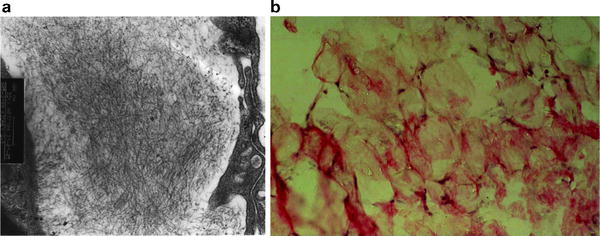
Fig. 37.3
Classic vascular purpura. (a) Electron micrograph demonstrating linear nonbranching fibrils of amyloid in a kidney biopsy specimen (original magnification × 100,000). (b) Congo red-stained fat aspiration specimen (original magnification × 1,000)
Although biopsy of the organ affected remains the diagnostic standard, many alternative methods are available [12]. Endoscopic biopsy of the upper digestive tract, even in the absence of gastrointestinal symptoms, may be highly sensitive for the diagnosis of renal amyloidosis. Amyloid has been reported [13] in the duodenum in 100 % of biopsies, in the stomach in 95 %, in the rectum in 91 %, and in the esophagus in 72 %. Endoscopy has clear advantages over renal biopsy because it is better tolerated, it can be performed on an outpatient basis, and the risk of hemorrhage after the biopsy is low [14]. Rectal, skin, and labial salivary gland biopsy can be performed. Labial salivary gland biopsy is minimally invasive and has been used to diagnose amyloidosis in patients presenting with a polyneuropathy [15]. When small amyloid deposits are anticipated, renal biopsy may be preferred because tissue is suitable for electron microscopic studies and for immunohistochemical classification [16].
The most commonly used technique to establish the diagnosis of amyloidosis is subcutaneous fat aspiration. The test yielded sensitivity of 84 % in a blinded study [17, 18]. It is considered the diagnostic procedure of choice because it requires little technical expertise and causes virtually no patient discomfort. The specificity of the test is 99 %, with 1 % false-positive results related to overstaining. The diagnosis can be established in 24 h. Concordance between two pathologists is 95 %. It is wise to interpret equivocally positive stains as being negative because the consequences of a false-positive interpretation can be extreme. A Congo red-stained fat aspirate is shown in Fig. 37.3b.
Methods exist to identify and classify amyloidosis from fat biopsy specimens. An enzyme-linked immunosorbent assay (ELISA) successfully characterized the type of amyloidosis in 14 of 15 positive fat biopsy specimens [19]. A second study using a similar technique showed a specificity of 100 % and a positive predictive value of 100 % [20]. The sensitivity in this study, however, was low at 58 %, and the percentage of inadequate specimens was quite high at 11 %. Ambiguous or equivocal Congo red stains due to pale staining of amyloid fibrils or birefringence of collagen in fat tissue contributed to the low sensitivity. Congo red fluorescence has been used to increase the sensitivity of the fat aspirate and can be used on archival tissues [21]. Analysis of the fat has also been used to characterize the type of amyloid, even when the presenting syndrome is in the myocardium [22]. Western blot analysis with specific antibodies to amyloid fibril proteins has also been used to establish the type of amyloidosis in subcutaneous fat [23]. In a study of 91 patients who underwent fine-needle aspiration of fat at a large academic medical center, the findings were positive in 22 %, negative in 68 %, insufficient in 9 %, and equivocal in 1 % [24]. Of the 62 patients who had negative fine-needle aspiration studies, follow-up biopsies were performed in 19, and 5 specimens were positive for amyloidosis when stained with Congo red. The sensitivity and specificity for fine-needle aspiration were 75 % and 92 %, respectively. A positive result for amyloid excluded other underlying conditions. In a study of 120 patients with established systemic amyloidosis (38 AA, 70 AL, and 12 transthyretin [ATTR]), all underwent fat biopsy. Smears were positive for amyloid in 93 % of patients [5]. The specificity was 100 % in 45 control-blinded samples, and fat tissue aspiration was superior to rectal biopsy. In an interpretation of the subcutaneous fat aspirate when one pathologist examined three smears, the sensitivity was 80 % but increased to 90 % when two pathologists examined three smears. The additive value of a subsequent rectal biopsy was negligible, so if the fat aspiration sample tests negative, biopsy of the affected organ is recommended.
A highly specific and sensitive novel test for the typing of amyloidosis in routine clinical biopsy specimens has been developed [25]. This approach combines specific sampling by laser microdissection (LMD) and tandem mass spectrometry (MS)-based proteomic analysis. The report studied 50 cases of amyloidosis that were well characterized by gold standard clinicopathologic criteria (training set) and an independent validation set comprising 41 cases of cardiac amyloidosis. By use of LMD MS, the amyloid type was identified with 100 % specificity and sensitivity. Accurate typing of the amyloid deposits is essential for accurate planning of therapy [26].
In summary, the preferred technique for the diagnosis of suspected systemic amyloidosis is subcutaneous fat biopsy. Specimens should be examined with Congo red. The bone marrow is positive in 50 % of patients. This diagnostic test is often required since the patients have a monoclonal gammopathy, and multiple myeloma must be excluded. At Mayo Clinic, the subcutaneous fat aspiration and bone marrow biopsy are usually done as a single procedure with the patients sequentially prone and then supine. In our hands, the combined use of this technique demonstrates deposits of amyloid in 87 % of patients.
Distinguishing Between Localized and Systemic Amyloidosis
Once a diagnosis of amyloidosis is established with a positive Congo red stain, two key questions remain before the patient’s prognosis can be assessed and appropriate therapy determined. The first question when amyloid deposits are found in tissue is whether the amyloidosis is localized or systemic. Figure 37.4 shows the distribution of amyloidosis types seen at Mayo Clinic. Patients with localized amyloidosis may present with symptoms that mimic systemic diseases. This can include hematuria, respiratory difficulty, and visual disturbances. Localized amyloidosis may often be suspected by the pattern of deposition. One common form of localized amyloidosis involves the genitourinary tract and can include the bladder, renal pelvis, or ureter [27]. These patients often present with obstruction, and the preoperative diagnosis is usually transitional cell carcinoma of the urothelium. Often this diagnosis is established after nephroureterectomy that, in retrospect, was unnecessary [28]. Localized amyloidosis is also typically found in the bladder. The presenting symptom is hematuria with urgency. Cystoscopy usually reveals a mass, and the preoperative diagnosis is transitional cell carcinoma of the bladder [29]. These patients are never treated systemically and are not found to have a systemic light chain disorder. A second typical site for localized amyloidosis is the larynx and tracheobronchial tree [30]. These patients present with hoarseness [31]. However, in rare instances, advanced involvement of the tracheobronchial tree can cause serious airflow obstruction [32]. It may also cause recurrent pneumonitis. Typically, the computed tomographic (CT) appearance is normal, and the diagnosis is established by biopsy during either bronchoscopy or laryngoscopy. The treatment is localized, usually involving laser vaporization of the amyloid deposits [33, 34].
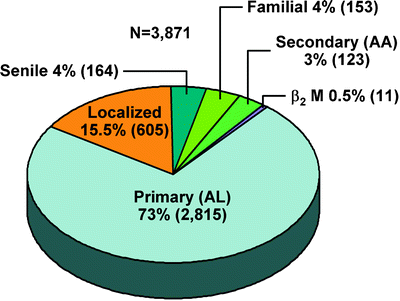
Fig. 37.4
Distribution of amyloid types seen at Mayo Clinic, 1960–2009
Cutaneous deposits of amyloidosis may be associated with AL amyloidosis as part of systemic light chain deposition, but more often the deposit is localized and analysis reveals the amyloid is composed of degenerated keratin protein misfolding into fibrils [35, 36]. It is not uncommon to find amyloidosis localized to the conjunctiva. Management is surgical [37], and preoperatively this presentation of amyloidosis can be confused with conjunctival lymphoma.
Patients may present to a physician with a localized pulmonary nodule. The prebiopsy diagnosis is generally malignancy. Biopsy shows pure amyloid deposits. This represents nodular pulmonary amyloidosis, which is an indolent condition. Although the nodule may progress over time, excision is not mandatory, and it does not reflect the presence of a systemic amyloid process [38]. Carpal tunnel syndrome is observed in 15 % of patients with amyloidosis, but the majority of patients who present with carpal tunnel syndrome whose carpal tissue stains positively with Congo red have a localized form of amyloidosis composed of transthyretin [39]. Trace amounts of amyloid can be seen in the cartilage of the hip and in the knee after resection and represent incidental findings that are not reflective of a systemic deposition of amyloid [40, 41]. A diagnostic algorithm for AL amyloidosis is found in Table 37.1.
Table 37.1
Diagnostic pathway for AL amyloidosis
1.Consider AL amyloidosis in patients with •Nondiabetic nephrotic syndrome •Nonischemic cardiomyopathy with an echocardiogram showing “hypertrophy” •Hepatomegaly or alkaline phosphatase elevation without imaging abnormality •Peripheral neuropathy with MGUS or CIDP with autonomic features •Atypical myeloma with monoclonal light chains and modest marrow plasmacytosis. |
2.Screen suspicious patients with electrophoresis and immunofixation of serum and urine and serum free light chain assay. If negative, inquire about the possibility of familial or localized amyloidosis. |
3.Biopsy patients with a monoclonal protein; fat and bone marrow should be sampled first. |
4.Assess prognosis with echocardiography including Doppler and strain echocardiography, evaluate serum troponin and NT-proBNP levels. |
5.Refer for anti-plasma cell therapy. |
Classification of Amyloidosis on Biopsy Tissue Specimens
Once the first question of localized vs. systemic amyloidosis has been answered, one can move on to the second question: whether systemic amyloidosis is light chain derived. Patients who have localized amyloidosis do not require extensive evaluation. Most patients require no therapeutic intervention, and when treatment is required, it is often limited to the specific organ of deposition. However, if amyloidosis is systemic, usually involving a visceral organ such as the heart, kidney, liver, peripheral or autonomic nervous system, gastrointestinal tract, or periarticular soft tissues, including the tongue, then the second question applies [42]. Figure 37.5 lists the clinical syndromes associated with systemic AL amyloidosis. Table 37.2 lists the forms of systemic amyloidosis that have been identified to date. Only immunoglobulin light chain amyloidosis (AL amyloidosis), the most common type, is treated with systemic chemotherapy because of its plasma cell origin. All other forms require alternate interventions, and chemotherapy is strictly contraindicated. Figure 37.4 gives the distribution of the forms of systemic amyloidosis seen at Mayo Clinic. The treatments for light chain, secondary, inherited, dialysis-dependent, and senile systemic amyloidosis vary dramatically, and the literature reflects reports of patients having been classified incorrectly and treated incorrectly. In one study, fibrinogen A α chain renal amyloidosis and ATTR amyloidosis were confirmed by genetic testing even though the original clinical diagnosis was AL amyloidosis [43]. Since monoclonal gammopathy of undetermined significance is found in 3–5 % of adult patients [44], the finding of amyloidosis and an associated monoclonal protein does not guarantee the amyloidosis is light chain in origin. Case reports of hereditary systemic amyloidosis with an incidental monoclonal gammopathy have been published [45]. The danger is that patients with amyloid deposits and a monoclonal gammopathy may be referred for chemotherapy treatment, which has no role in hereditary amyloidosis. One reported patient was found to have nephritic-range proteinuria and an IgM monoclonal protein associated with Waldenström macroglobulinemia and was thought to have amyloidosis but ultimately was found to have minimal change disease [46]. Challenges inherent in the determination of the type of amyloid from a biopsy specimen arise because of the small tissue sample and the frequent finding of trace amounts of Congo red-positive materials in a biopsy specimen. Microscopic methods have been developed for the extraction, purification, and amino acid sequencing of amyloid proteins contained in minute specimens. Amyloid proteins may be extracted in sequence from formalin-fixed tissue specimens. An exact identification of the protein in deposits is possible [47]. Amyloid proteins that are purified by microextraction techniques can then be subjected to immunochemical and direct chemical characterization [47]. Monoclonal antibodies have been developed that can immunostain pathologic light chain-related amyloid and nonfibrillar tissue deposits [48].
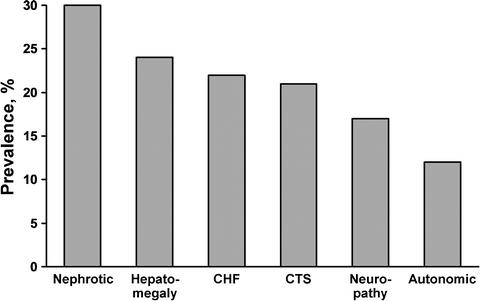
Fig. 37.5
Prevalence of clinical syndromes in AL amyloidosis. CHF congestive heart failure, CTS carpal tunnel syndrome. (Data from Kyle RA, Gertz MA. Systemic amyloidosis. Crit Rev Oncol Hematol. 1990;10(1):49–87.)
Table 37.2
Classification of systemic amyloidosis
Abbreviation | Protein subunit | Clinical characteristics |
|---|---|---|
AL | Immunoglobulin light chain | Cardiomyopathy, nephrotic syndrome, hepatomegaly, neuropathy |
AH | Immunoglobulin heavy chain | Same as AL |
ATTR | Native transthyretin (senile systemic amyloidosis) Mutant transthyretin (familial amyloidosis) | Cardiomyopathy, carpal tunnel Polyneuropathy, cardiomyopathy |
A Fib | Variant fibrogen A α chain (inherited) | Nephropathy |
A Apo AI | Mutant apolipoprotein AI familial | Polyneuropathy |
ALys | Mutant lysozyme | Hepatopathy |
AA | Amyloid A protein | Nephropathy, enteropathy goiter |
Reactive | Chronic rheumatic or infectious | |
Inherited | Familial periodic fever syndrome, including familial Mediterranean fever | |
A β 2 M | β2-Microglobulin (dialysis amyloid) | Arthropathy, carpal tunnel syndrome |
Analytic techniques that have been used to identify amyloid deposits include ELISA, Western blot, direct amino acid sequencing, and mass spectroscopy [49, 50]. Immunohistochemical assays are used most commonly, although controversy exists regarding the sensitivity of the technique [6]. In an autopsy series of 43 patients, noncommercial antibodies were used and identified AA in 21, ATTR in 11, and AL in 10. One patient had more than one type of systemic deposition. With AA and AL amyloidosis, the kidney was involved most frequently. In ATTR amyloidosis, the heart and lungs were involved most frequently. The amyloidosis could be classified using specific antibodies against five major amyloid fibril proteins. Microextracts of amyloid proteins from 11 fat aspirates were analyzed immunohistochemically with concordant results in three of four with κ amyloid, five of six with λ amyloid, and one with AA [18, 51]. Polyclonal antibodies against synthetic amyloid peptides have been developed to positions 118–134 of λ light chains and positions 116–133 of κ light chains. These synthetic peptides can be used to classify amyloid in paraffin-embedded tissue specimens [52]. In this study, anti-λ antiserum reacted with samples from 18 of 19 λ amyloids, and anti-κ antiserum reacted with 9 of 10 κ amyloids. Immunofluorescence staining of kidney biopsy specimens has been demonstrated to be unreliable. One study described negative immunofluorescence results for 12 patients with plasma cell dyscrasias representing more than a third of study patients [10]. Because the sensitivity of immunofluorescence microscopy in the kidney was low, additional diagnostic studies were required. In a series of 169 biopsies performed on 121 patients, only 12 specimens could not be classified using immunohistochemistry [53]. In 32 % of biopsy specimens, amyloid deposits were equivocally stained for κ or λ, reflecting the limited sensitivity of immunohistochemical analysis for light chain amyloid. There are two possible explanations for this. In many amyloid deposits, only a fragment of the light chain is deposited, and it may not be possible for antibodies, which may be directed at the constant region of the light chain that could have missing epitopes, to bind to the light chain protein. A second possibility is that when amyloid misfolds into the β-pleated sheet configuration, previously exposed epitopes are buried into the interior of the configured fibril where they are no longer exposed to the antiserum. Immunohistochemical classification poses a problem, which is magnified when commercial antibodies are used. The use of commercial antibodies for detection of AA and ATTR amyloidosis is relatively straightforward. Unfortunately, the same cannot be said for trying to recognize AL amyloid deposits in paraffin-embedded tissues [53, 54]. Patients with ATTR amyloidosis often lack a family history, and lack of a family history cannot be used as an exclusionary criterion for the diagnosis. Cardiac ATTR amyloidosis has superior survival compared with AL amyloidosis and is not treated with chemotherapy, so the distinction is clinically important [55].
Mistakes that are commonly cataloged as resulting in misdiagnosis of the type of amyloidosis include inconsistent immunolabeling, nonspecific background staining, and inconsistent chemical reactions [6]. Newly developed antibodies specifically stain proteins in Western blots and formalin-fixed paraffin-embedded tissue sections [56]. Formic acid extraction has been added to immunochemical and biochemical characterization to enhance the exposure of epitopes to the antibodies. The exposure of constant region sequences of the light chain increases diagnostic sensitivity [49].
Mass spectrometry is the newest technique available for the diagnosis of systemic amyloidosis. A microtechnique can be used to extract fibrillar deposits from formalin-fixed biopsy specimens. The chemical composition of the deposits can then be determined through direct amino acid sequencing [57, 58]. This technique has allowed identification in tissue section of pure heavy chain amyloid with no light chain circulating in serum as well as mixed forms of amyloid containing both light chain and transthyretin proteins [59].
A unique molecular profile in light chain amyloid has been identified through functional gene expression analysis of clonal plasma cells [60]. Class prediction analysis shows a subset of 12 genes that can be used to discriminate AL amyloid from other amyloid protein. Molecular profiling of clonal plasma cells may provide insights into the pathogenesis of AL amyloidosis.
In summary, after amyloidosis has been diagnosed, it is essential to classify its type from tissue biopsy specimens. The recently introduced immunoglobulin free light chain assay [61] has been used to further classify the type of amyloidosis. Abnormalities of the immunoglobulin free light chain ratio may be specific for AL amyloidosis, but an absence of monoclonal free light chains has been reported and can result in false-negative findings [62]. Antiamyloid antibodies have been found in pooled human immunoglobulin that can recognize the fibrillar confirmation associated with all forms of amyloidosis, including AA, ATTR, and islet amyloidosis. These antibodies immunostain amyloid tissue deposits and have potential as a diagnostic agent for patients with amyloid-associated disease [63].
Imaging
Imaging of Amyloid Deposits Using Radionuclides
Radionuclide imaging has been attempted to identify amyloid deposits. Technetium 99m-labeled pyrophosphate was used in an attempt to identify myocardial amyloidosis [64]. The technique did not demonstrate notable tracer uptake but was able to recognize a reduction in systolic function and impairment of diastolic function. Sporadic case reports have described the use of technetium-labeled pyrophosphate for diagnosing amyloid polyneuropathy [65, 66].
Thallium 201 scintigraphic studies have been performed in patients with cardiac amyloidosis. Washout rates during rest and during delayed thallium imaging may reflect the severity of amyloidosis in the heart, and mean washout rates are higher in patients with amyloidosis than controls. In four of five patients with amyloidosis, the washout rate of thallium was high, and all patients studied succumbed to the disease in less than a year [67].
Gallium 67 and thallium 201 single-photon emission computed tomography (SPECT) imaging has been used to study dialysis amyloidosis. This technique was compared to imaging with technetium-labeled methylene diphosphonate. Technetium-labeled methylene diphosphonate whole-body bone scans are able to detect active and preexisting deposits of dialysis amyloid. Gallium and thallium scans were helpful in differentiating between active and inactive deposits when evaluating the therapeutic effect of intervention [68]. Technetium-labeled N2S2 conjugates of chrysamine G have been used to image amyloid deposits. This agent localizes specifically to amyloid deposits in human kidney tissues and can potentially act as a diagnostic targeting agent [69]. The use of technetium 99m-3,3-diphosphono-1,2-propanodicarboxylic acid in eight patients with ATTR amyloidosis was highly sensitive, showing whole-body tracer retention of 80 % in patients with familial amyloidotic polyneuropathy compared with 56 % in controls 3 h after injection [70]. Increased uptake of technetium-labeled bone tracer hydroxymethylene diphosphonate has been reported for patients with cardiac amyloidosis [71].
Technetium-labeled aprotinin [72, 73] also has been studied as a specific marker of amyloidosis because it appears to be a sensitive diagnostic molecule. In 23 patients, 22 had focal accumulations of technetium-labeled aprotinin in different organs. Findings for 20 of these patients were confirmed at autopsy or through biopsy. The diagnostic accuracy of technetium 99m-3,3-diphosphono-1,2-propanodicarboxylic acid was investigated in cardiac amyloidosis [74, 75]. The accuracy for differentiating ATTR from AL deposits of amyloid was 100 %, and there was no uptake in controls. The sensitivity and specificity in ATTR amyloid were 100 %. In AL patients, the sensitivity was 0 % and the specificity was 100 %. Therefore, technetium 99m-3,3-diphosphono-1,2-propanodicarboxylic acid was useful for differentiating ATTR and AL amyloid.
On an experimental basis, high-resolution microradiographic imaging of amyloid deposits has been developed in a murine model. It combines radiolabeled serum amyloid P (SAP) component and SPECT imaging. The use of radiography to discern the extent of the amyloid burden was useful in quantifying the total body burden of amyloid and for evaluation of the therapeutic efficacy of new pharmacologic agents [76]. Radioimaging of light chain amyloid with a fibril-reactive monoclonal antibody has been studied in mice. The radioiodinated antibody retains immunoreactivity localized in the mouse tumors and was easily visible in SPECT/CT [77].
The SAP Component Scan
The radioiodinated SAP component is a highly specific tracer molecule that identifies amyloid deposits. The SAP component is a nonfibrillar pentraxin plasma protein that binds specifically through a calcium-dependent mechanism to amyloid fibrils and is always present in amyloid deposits. Twenty years ago, scintigraphy with iodine 123-labeled SAP was first used to diagnose, locate, and monitor the extent of systemic amyloidosis [78]. The uptake of the radionuclide is proportional to the quantity of amyloid protein in the different tissues, and 24-h retention levels are abnormal in all patients with considerable total body burdens of AL amyloid [79]. The serialized SAP component scan has been used to show the regression of visceral amyloid deposits after liver transplant in patients with ATTR amyloidosis [80]. With this technique, hepatic amyloid deposits were identified in 54 % of AL patients and 18 % of AA patients but only 2 % of patients with familial amyloidosis [81]. The results of SAP scans are concordant with liver biopsy findings. On the basis of the scan, liver involvement is always associated with amyloid deposits in other organ systems. The scan is used as a noninvasive method for monitoring patients after kidney transplant for the possibility of disease recurrence [82]. Radiolabeled iodine 131 has also been used, but its imaging characteristics are unfavorable compared with iodine 123, the preferred radioisotope. This scan is not widely available and does not image the myocardium so other methods are required to detect cardiac involvement.
Systemic amyloidosis is characterized by accelerated clearance from plasma of iodine 123-labeled SAP. It is also characterized by an increased interstitial exchange rate and extravascular retention. These findings indicate that the binding of radiolabeled SAP is reversible and that SAP imaging can be used to monitor the effects of therapy and demonstrate regression of amyloid deposits [4, 83]. The scans provide direct imaging of organ distribution of the amyloid deposits, proof of amyloid in sites unavailable to biopsy, and demonstration of progression or regression of the deposits in various organs.
Echocardiographic Assessment of Amyloidosis in the Myocardium
Echocardiography is a powerful tool used to establish the diagnosis of cardiac amyloidosis and to differentiate cardiac amyloidosis from hypertrophic cardiomyopathy [84]. The echocardiographic features in amyloidosis are well described and include left ventricular fractional shortening, a transmitral flow velocity compatible with abnormal relaxation, right ventricular systolic dysfunction, and ventricular wall thickening [85]. Pulse tissue Doppler imaging adds value to two-dimensional imaging studies for evaluation of systolic and diastolic left ventricular function [86]. Right ventricular dysfunction in cardiac amyloidosis has been characterized using the Tei index [87]. Cardiac amyloidosis may be diagnosed using the velocity profile in the hypertrophied left ventricular wall. The myocardial velocity profile in the septum and left ventricular posterior wall shows a distinctive serrated pattern that may be related to amyloid deposition in the myocardium. Color-coded tissue Doppler imaging also provides diagnostic information [88]. Low-voltage pattern, pseudoinfarction patterns, increased myocardial thickness, and a speckled appearance of the myocardium on the echocardiogram are associated with biopsy-proven cardiac amyloidosis. Combining electrocardiography, echocardiography with strain, and general indications for myocardial biopsies with correct analyses is needed to diagnose cardiac amyloidosis [89]. A combination of low voltage and myocardial thickness can be used as a powerful statistical predictor of mortality [90]. Infiltration of the myocardium may occur in any form of systemic amyloidosis, but it is quite rare in AA amyloidosis until late in the disease or years into dialysis therapy. Echocardiography is useful in detecting amyloidosis in this rare subgroup [91]. The development of strain rate imaging to assess myocardial function has added considerably to tissue Doppler evaluation of patients with cardiac amyloidosis. It detects the earliest degrees of cardiac dysfunction and is now a routine part of assessment of patients with cardiac amyloidosis at Mayo Clinic [92–94]. The use of strain imaging can differentiate patients with cardiac amyloidosis from those without cardiac involvement, including cardiac patients who have normal wall thickness [95].
Magnetic Resonance Imaging Assessment of Cardiac Amyloidosis
Magnetic resonance imaging (MRI) is a noninvasive diagnostic method that can be used to specifically identify amyloid deposits in the heart. Delayed enhancement on MRI following gadolinium infusion occurs in 69 % of patients with cardiac amyloidosis. The delayed enhancement is seen in the subendocardial layer. Diffuse myocardial enhancement may also be seen in some [96]. The delayed enhancement may suggest interstitial fibril expansion with associated endomyocardial fibrosis. Screening of subclinical early cardiac involvement may become possible. Myocardial enhancement is associated with increased ventricular mass and impaired left ventricular systolic function [97]. Patients with cardiac amyloidosis have a characteristic pattern of global, subendocardial late enhancement coupled with abnormal gadolinium kinetics in both the myocardium and blood pool, which fits with the transmural histologic distribution of amyloid. Late gadolinium enhancement is common in cardiac amyloidosis. Global transmural or subendocardial gadolinium enhancement is most common, but suboptimal myocardial nulling and focal patchy gadolinium enhancement are also observed. Gadolinium enhancement may detect early cardiac abnormalities in patients with amyloidosis with normal left ventricular thickness. The presence and pattern of gadolinium enhancement are strongly associated with clinical, morphologic, functional, and biochemical markers of prognosis [98].
MRI has also been useful for the diagnosis of amyloid arthropathy, identifying lesions in joint capsules and tendon, and tumor-forming periarticular and osseous lesions. Both these findings can be useful for amyloid arthropathy [99].
In summary, all patients with amyloidosis must have Congo red stain-positive deposits showing apple-green birefringence. Subcutaneous fat is the least invasive highly sensitive source of tissue. Either the fat aspirate or marrow biopsy specimen reveals deposits of amyloid in most patients (Fig. 37.6). The amyloid deposits must be characterized to establish whether the process is localized or systemic, and all forms of systemic amyloidosis must be further classified to distinguish among the various forms. Echocardiography and MRI are important tools for assessing the extent of cardiac amyloidosis.
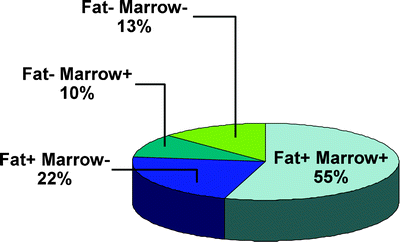
Fig. 37.6
Frequency of positive findings of amyloid in fat and bone marrow specimens
Pathophysiology and Prognosis
The actual mechanism whereby amyloid deposits produce organ dysfunction remains unclear. Whether the finely formed amyloid deposit is responsible for organ dysfunction or whether there are soluble toxic intermediates that mediate this dysfunction remains unclear. In ATTR amyloidosis, soluble aggregates were found not to be toxic, but monomeric ATTR or small aggregates not exceeding six subunits were the major cytotoxic species. Therefore, it may not be the final product of amyloid that is responsible for tissue damage, but rather aggregates that form low-molecular-mass assemblies that are highly cytotoxic [100]. This hypothesis fits with the fact that there is a poor correlation between the extent of amyloid deposits and the amount of organ dysfunction. The soluble toxic intermediates may exert tissue toxic effects independent of the amount of amyloid deposited [101].
In theory, there are two mechanisms of interference with the deposition of amyloid deposits. One would be to interfere with the misfolding of the fibril, rendering it soluble or stabilizing it in a monomeric state. Unfortunately, in AL amyloidosis, there are no published studies in humans that attempt to solubilize the amyloid. It is known, however, that amyloid deposits are in dynamic equilibrium with the soluble pool, and if the precursor protein production is interrupted, these deposits can regress. The equilibrium constant varies from light chain to light chain, and how much reduction is necessary for amyloid deposits to resolubilize remains unknown and cannot be measured. The variable equilibrium of each patient’s light chain makes it difficult to assess the goal of therapy, e.g., 50 % reduction, 90 % reduction, or complete eradication of light chains. All current strategies to manage amyloidosis involve cytotoxic therapies designed to destroy the plasma cell responsible for synthesis of the immunoglobulin light chain. The types of monoclonal immunoglobulins seen in the serum of AL patients are shown in Fig. 37.7 and are contrasted to the proteins found in myeloma to highlight the higher prevalence of light chain proteinemia and λ over κ when compared with myeloma. Virtually every therapy that has been attempted in the management of amyloidosis has been derived from favorable results obtained in patients with multiple myeloma. Generally, efficacy has first been demonstrated in multiple myeloma patients and is subsequently applied to amyloidosis patients. It has been 30 years since the first published report of alkylating agent-based chemotherapy to treat amyloidosis [102, 103].
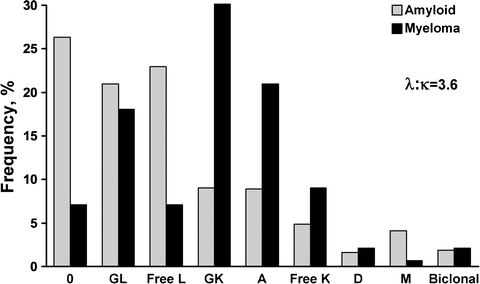
Fig. 37.7
Comparison of serum immunofixation results immunoglobulins in myeloma and amyloidosis
When a patient presents with a syndrome compatible with amyloidosis (Fig. 37.5) and is found to have a monoclonal protein and the diagnosis is confirmed histologically, the next step is to assess the patient’s prognosis. The cause of death in most patients with AL amyloidosis is cardiac, either cardiomyopathy that progresses to heart failure or sudden death caused by ventricular fibrillation or asystole [104]. Congestive heart failure is the most powerful indicator of survival. Syncope, likewise, is a powerful predictor of sudden death. Measures of myocardial performance by echocardiography, such as the Doppler-derived index of diastolic performance, are important in prognosis. A deceleration time of 150 ms or less on Doppler echocardiography indicates restrictive physiology and a lower percentage of 1-year survival [105]. The level of serum β2-microglobulin and the difference between involved and uninvolved immunoglobulin light chain are predictive for overall survival. The four most powerful factors are the difference between serum levels of the normal involved and uninvolved free light chains and the serum levels of troponin T, the N-terminal fragment of pro-B-type natriuretic peptide (NT-proBNP), and β2-microglobulin. The weakest of those four variables is NT-proBNP, and significant survival prediction can be achieved using the serum levels of free light chain, troponin T, and β2-microglobulin. Baseline free light chain correlates with serum cardiac troponin levels. Higher free light chain levels are associated with more organs involved by amyloid, and the absolute level of free light chain achieved after therapy correlates with survival [106].
To interpret the value of various therapeutic options available for patients with AL amyloidosis, the response to therapy in this disorder must be clearly defined. In multiple myeloma, the majority of patients have a quantifiable M protein that can be used as a surrogate for reduction of tumor mass. Interpreting a response in a patient with AL amyloidosis is far more complex. The majority of AL patients do not have an intact immunoglobulin protein that is measurable. The majority of patients have only light chain disease so until recently quantification has been challenging. Only 10 % of AL patients have an M protein level higher than 1.5 g/dL. Quantification of the urinary light chain is equally difficult because a high proportion of patients have albuminuria, which makes it impossible to visualize and measure an M component in the urine. Bone marrow assessment is unreliable, because patients with AL amyloidosis at diagnosis average only 5–6 % clonal plasma cells in the bone marrow, and detection of a reduction in bone marrow plasmacytosis as a measure of response is subject to sample variability and interpretive concordance. Previously, a hematologic response for amyloidosis had been defined by a 50 % reduction in the precursor monoclonal protein [107]. This definition, however, is severely lacking because it does not incorporate the critically important role of the immunoglobulin free light chain assay in assessing for response in AL amyloidosis. Not only is the free light chain level an important clue for classifying the amyloidosis as light chain in origin, it provides a new method for confirming a hematologic response [108]. It is well recognized that decreases in free light chain are more readily detected early after treatment than are changes in serum and urine immunofixation and bone marrow studies [109] and that reduction in the free light chain is concordant with improvement with cardiac biomarkers and predicts improved survival. Patients in whom chemotherapy fails to induce a decrease in free light chains are at risk of early death [110]. An abnormal free light chain assay is found in virtually 100 % of AL patients at the time of diagnosis [111]. When free light chain levels normalize, histologic regression of amyloid deposition in fat tissue has been demonstrated. Following chemotherapy, reduction of two grades of amyloid deposition in fat tissue was seen in 50 % of patients after 2.4 years and 80 % after 3.2 years. The best histologic responses were seen in patients with complete normalization of the free light chain [112]. Controversy remains as to whether the goal of treatment should be a 50 % reduction in free light chain or whether attempts to consolidate patients to achieve a complete response should be undertaken [113]. It has been suggested by some that changes in the intact immunoglobulin level are of far less utility in predicting outcome than changes in the serum free light chain level. In one cohort, the best predictor of long-term survival was a more than 90 % reduction in the difference between involved and uninvolved free light chain value. Part of the reason that response rates appear to have improved from the 1970s to the current era is the ability to regularly and accurately measure serum free light chain levels and reproducibly demonstrate a quantifiable reduction.
Conventional Treatment of Amyloidosis
Alkylating Agent-Based Chemotherapy
Shortly after the introduction of the melphalan–prednisone combination for the treatment of multiple myeloma, it was demonstrated to be an effective therapy in amyloidosis [114, 115]. Disadvantages of alkylator-based therapy include a low response rate and a notable delay between the onset of therapy and the demonstration of a hematologic response. These delays make it difficult to distinguish whether lack of improvement in hematologic parameters reflects a therapeutic failure or a delay in development of a response. The estimated median time for evidence of response from melphalan–prednisone treatment is approximately 1 year. Oral melphalan therapy can also result in late myelodysplasia. When the duration of melphalan treatment often extended to 2 years, nearly 7 % of exposed patients developed myelodysplasia [116]. As the median survival of amyloidosis has been steadily improving as a result of better available therapies and improved supportive care, the number of patients at risk for developing myelodysplasia actually has been increasing, with an actuarial risk at 10 years of 20 % [117]. Reported response rates to melphalan–prednisone are approximately 30 % [103]. Immunoglobulin free light chain assay was not available in that era, so the response rate may be an underestimate. The key advantage is that melphalan–prednisone is so well tolerated that it can be administered to virtually any patient with AL amyloidosis regardless of performance status, serum creatinine level, or extent of cardiac failure. If a patient is willing to undergo a trial of melphalan–prednisone and understands the risks, there is no good reason to withhold therapy [118].
The best responses seen to melphalan–prednisone occur in patients with single-organ nephrotic syndrome with maintained serum creatinine levels. Using strict response criteria, the overall response rate to melphalan–prednisone is 18 % [119]. If the patient’s alkaline phosphatase level is four times normal or the serum creatinine level is higher than 3 mg/dL, a response to melphalan–prednisone is rare, and the likelihood of developing dialysis-dependent renal failure is high. Clinically important responses have been reported with melphalan–prednisone therapy even in the presence of advanced cardiomyopathy, and in a prospective randomized trial, a modest prolongation of survival has been demonstrated in the presence of amyloid heart failure [120]. In a retrospective analysis of 153 patients treated with melphalan–prednisone, the 5-year survival of responders was 78 % [119]. In a study of 810 patients, the 10-year survival was 4.7 %, and all long-term survivors had been treated with melphalan–prednisone [121]. Fourteen of the 30 10-year survivors had a complete hematologic response to melphalan–prednisone therapy. Two prospective randomized studies have demonstrated a survival advantage with melphalan-based therapy. One three-arm study accrued 219 patients and randomly assigned them to receive colchicine, 0.6 mg twice daily, standard melphalan–prednisone, or melphalan plus prednisone and colchicine. Half the patients had proteinuria in the nephrotic range, and 20 % had overt clinical heart failure. The median survival for both melphalan-containing regimens was 17 months compared with 8.5 months with colchicine alone [122]. A second prospective randomized study of 100 patients, 50 receiving colchicine and 50 receiving melphalan–prednisone and colchicine, showed a median survival of 6.7 months in the colchicine group vs. 12 months in the three-drug group [123]. Since there is a clear survival advantage in the melphalan-based group, withholding therapy is not justified, but the short median survival reflects the need for therapeutic improvements.
In patients whose cardiac status makes anything other than the gentlest therapy impractical, continuous oral daily melphalan has been used. In a report of 30 such patients with AL amyloidosis, 7 of the 13 patients who were evaluable after 4 months of therapy achieved a partial hematologic response, and the response was complete in 3. This suggests melphalan can provide marked palliation, even in patients with far-advanced disease [124].
Dexamethasone-Based Regimens
Vincristine, doxorubicin (Adriamycin), and dexamethasone (VAD) therapy was reported in eight AL patients. Four fulfilled criteria for an objective hematologic response [125]. VAD is a suboptimal regimen in the management of amyloidosis because vincristine should not be used in patients with amyloid neuropathy, and doxorubicin is not a good choice because of the high prevalence of amyloid cardiomyopathy. When four patients with isolated amyloid nephrotic syndrome were treated with VAD, three responded and were alive and in remission anywhere from 4 to 9 years [126]. VAD has been used for pretransplant induction to reduce the plasma cell burden before collection of stem cells, but clinical improvement associated with VAD did not reduce transplant-related mortality [127]. Ninety-two patients received four cycles of VAD with an organ response in 39 of 92 (42 %) and treatment-related mortality of 7 % [107]. Nine consecutive patients with AL amyloidosis were treated with dexamethasone for 3–6 cycles at a dose of 40 mg on days 1–4, 9–12, and 17–20, followed by maintenance interferon, 3–6 million units three times per week [128]. Improvement in organ involvement was seen in eight of nine patients, and six of the seven patients with nephrotic syndrome had a 50 % reduction in albuminuria. We have reported on 44 patients with amyloidosis, a high proportion of whom had cardiac involvement [129, 130]. Ten percent have survived longer than 10 years. Reduced-frequency dexamethasone can result in response in up to 35 % of patients, with a median time to response of 4 months [131]. Ninety-three patients with AL amyloidosis were enrolled in a multicenter cooperative group trial of dexamethasone and interferon. Hematologic complete responses were seen in 24 % and improvement in organ dysfunction was seen in 45 % of evaluable patients, with a median survival of 31 months and a 2-year survival of 60 % [132]. Heart failure and elevated serum β2-microglobulin levels predicted adverse outcome. In a subset of patients who were eligible for stem cell transplant, the estimated 2-year overall survival was 78 %. The toxicity of full-dose dexamethasone in amyloidosis is substantial. Fluid retention is particularly problematic in patients with hypoalbuminemia secondary to nephrotic-range proteinuria and in those with restrictive cardiomyopathy. Dose modifications of dexamethasone are required. In the dexamethasone-interferon study, the 24 patients with cardiomyopathy had a significantly lower response rate to treatment. The median time to hematologic response was 103 days. Toxicity from dexamethasone was proportionate to the number of organs involved, and the only statistical factor that predicted toxicity was the presence of heart failure.
Melphalan with Dexamethasone: A Standard of Care
Use of melphalan plus high-dose dexamethasone has been reported by multiple groups for its utility in the management of AL amyloidosis. The majority of patients were selected for study on the basis of their ineligibility for high-dose melphalan followed by autologous stem cell transplant. In the first study of 46 patients, a hematologic response was seen in 31, and a hematologic complete response was seen in 15 (33 %). Improvement in organ function was seen in 22 patients (48 %). Advantages of this oral regimen include low day 100 all-cause mortality of 4 %, with resolution of symptomatic cardiac failure in 6 of 32 patients, a median time to response of 4.5 months, and only 11 % grade 3–4 adverse effects [133]. The results of this study were recently updated. The actuarial 6-year survival was 50 %, and progression-free survival was 40 % [134]. Patients previously treated who subsequently progress could be successfully retreated demonstrating a second response with melphalan and dexamethasone. Of 41 evaluable patients, there were 30 responders and 11 nonresponders. The median survival of responders has not been reached, and the median survival of nonresponders was just over 2 years. Boston University has used pulsed low-dose melphalan in patients ineligible for stem cell transplant because of either severe cardiac involvement or poor performance status [124]. The melphalan dose was adjusted to produce mid-cycle myelosuppression, and all patients received granulocyte growth factor support. Fifteen patients (median age, 55 years) received a median of three cycles. Eight of ten evaluable patients had a hematologic response, two of which were complete responses. Of the 15 patients treated, 2 survived 6 months; at 24 months after treatment, 13 had died. The median survival was 2 months, and ten patients died within 5 weeks of starting treatment. Not all the reports on the use of melphalan with dexamethasone for stem cell transplant-ineligible patients have been encouraging. A report from Memorial Hospital described treatment of 40 consecutive patients with oral melphalan and prednisone who had a median survival of only 10.5 months. Predictors of survival included sex, troponin levels, and intraventricular septal thickness. The hematologic response rate was 58 %; 13 % had complete responses. This poor outcome points out the importance of the patient population in interpreting single-arm, phase 2 reports [135].
One hundred forty-four patients in Great Britain received melphalan parenterally at a dose of 25 mg/m2 with dexamethasone, 20 mg, for 4 days every 21–28 days. The median number of cycles administered was 3. Fifty-one patients did not receive dexamethasone. The treatment-related mortality was 2 %; 23 % had normalization of free light chains, and 31 % had a 50 % decrease in serum free light chain. Forty-six percent were considered nonresponders. The response rate was higher in patients who received dexamethasone. Most clinical responses were evident within two cycles. Median survival for responders was 44 months, and amyloid-affected organ function improved in 14 % [136].
Thalidomide
Sixteen patients were enrolled in the first study of thalidomide for AL amyloidosis. The median maximum tolerated dose was 300 mg daily. Fifty percent of patients experienced grade 3–4 toxicity; 25 % had to discontinue thalidomide; 25 % had a reduction in light chain proteinuria but did not achieve an organ response manifest by reduced urinary albumin loss [137]. In a second report on 12 patients with AL amyloidosis who received thalidomide, 75 % had severe drug-related toxicity [138]. Progressive renal insufficiency was seen in 5, deep vein thrombosis in 2, and syncope in 2. The median time of thalidomide administration was only 72 days because of the high frequency of thalidomide intolerance. A third study of the use of thalidomide combined with dexamethasone reported 31 patients. Only 11 patients tolerated 400 mg per day, and the median duration of therapy was 5.7 months [139]. Twenty of the patients experienced grade 3 or greater toxicity. In an effort to reduce the toxicity, a lower dose of thalidomide was combined with melphalan and dexamethasone. This triple combination was administered to 22 patients with advanced cardiac amyloidosis. Eight hematologic responses and four organ responses were reported. Six patients succumbed to their advanced cardiac amyloidosis before cycle 3, which is a common outcome in patients whose diagnosis of cardiac amyloidosis is made late in the course of the disease [140].
In Great Britain, thalidomide was administered at a median dose of 100 mg per day and still required discontinuation in 31 % of patients. The hematologic response rate was 55 %. This regimen was combined with cyclophosphamide and dexamethasone. The 3-year estimated overall survival was 100 % and 82 % among complete and partial responders, respectively. The treatment-related mortality was 4 %, and the regimen could be used in poor-risk patients. Thalidomide does appear to be an active agent in amyloidosis, but the toxicity is substantial and the thalidomide dose is best started at 50 mg and should not exceed 100 mg. Symptomatic bradycardia is seen in as many as 26 % of patients. There have been a number of interesting individual case reports that validate the value of thalidomide. A 62-year-old man with New York Heart Association (NYHA) class IV heart failure was treated with thalidomide, 100 mg per day, with dexamethasone for 4 days every 4 weeks. The patient had a complete hematologic response at 8 weeks and an improvement in his cardiac failure to NYHA class II 2½ years after the start of therapy [141]. A second patient with advanced cardiac amyloidosis showed electrocardiographic improvement of low voltage and disappearance of proteinuria. Diastolic function showed remarkable improvement, and the patient was alive at 15 months and clinically stable [142].
Lenalidomide
Lenalidomide has been combined with dexamethasone in the treatment of AL amyloidosis in the hope that the toxicity typically associated with thalidomide would be reduced. Thirty-four patients with previously treated AL amyloidosis participated in a phase 2 trial with a starting dose of lenalidomide at 25 mg per day. This dose was not well tolerated and was reduced to 15 mg per day, which was the maximum tolerated dose. Of 24 evaluable patients, 7 (29 %) achieved a hematologic complete response and 9 (38 %) achieved a partial hematologic response for an overall response rate of 67 %. Hematologic responses were associated with organ responses. Fatigue and myelosuppression were the most common treatment-related adverse effects, seen in 35 %. Thromboembolic complications were seen in 9 % and were the most serious [143]. The cohort ultimately accrued 43 patients with a median follow-up of 26.5 months. Sixty percent had prior stem cell transplant, and 14 % had prior thalidomide. Lenalidomide, 15 mg per day, was given for 21 of 28 days. The median tolerated dose in the expanded cohort was 10 mg. The complete response rate was 24 % and the partial response rate was 36 %, with 15 % minor responses. Two of the complete responses were achieved late in the course of therapy at 18 and 19 months. Kaplan–Meier survival for 43 patients was 60 % at 3 years. Seven of the eight patients who achieved a complete hematologic response had renal amyloid nephrotic syndrome; in two the proteinuria resolved, and in three it declined by 50 %. The remissions were durable after therapy was discontinued.
Mayo Clinic conducted a trial of lenalidomide along with the addition of dexamethasone if patients did not achieve a hematologic response after three cycles. Twenty-three patients were enrolled in the first phase. Thirteen were previously treated. Sixty-four percent had cardiac involvement, and 73 % had renal involvement. Within the first three cycles of therapy, ten patients discontinued treatment. There were four early deaths, three adverse events, and three other causes for withdrawal. Some patients had more than 1 form of the disease. The most common grade 3–4 adverse events were neutropenia, thrombocytopenia, rash, and fatigue. When the cohort expanded to 37 patients, the starting dose of lenalidomide was reduced to 15 mg for the final 15 patients placed on the study. Only 5 of the original 37 patients were still on therapy at the time the report was published. The overall hematologic response rate was 43 %, but there was only one complete response and only one patient responded to lenalidomide without the addition of dexamethasone. The overall organ response rate was 20 % [144]. Thirteen patients were unable to complete 3 months of therapy. The major predictor of the inability to complete 3 months of therapy was cardiac biomarkers at the start of treatment, troponin T and NT-proBNP levels. Thus, if the 13 patients who were inevaluable for response were excluded from analysis, i.e., those who could not complete 3 months of therapy and, therefore, never had dexamethasone added to lenalidomide, the hematologic and organ response rates were 62 % and 32 %, respectively. The median response duration, progression-free survival, and overall survival were 14.8, 18.8, and 31 months, respectively. Overall survival from the original AL diagnosis was 48 months. Patients with a troponin T level higher than 0.035 ng/mL and an NT-proBNP level higher than 332 pg/mL were more likely to drop out early and less likely to respond. Sixty percent of patients with elevated troponin and BNP dropped out by cycle 4, reflecting the difficulty in administering any form of oral therapy to high-risk patients. Progression-free survival stratified by the Mayo Clinic biomarker staging system for stages 1, 2, and 3 were 34.7, 18.3, and 4.4 months, respectively [145].
Forty patients with relapsed or refractory AL amyloidosis were treated with lenalidomide and dexamethasone. Lenalidomide was initiated at 15 mg daily and was reduced to every other day if the creatinine clearance was less than 40 mL/min and to three times per week for patients on dialysis. Dexamethasone, 20 mg, was given for four consecutive days every 28 days. Prophylaxis against venous thromboembolism was given in the form of aspirin or low-molecular-weight heparin; 19 had prior stem cell transplant; 21 were previously treated with conventional-dose melphalan-based chemotherapy. The median time from diagnosis to initiation of lenalidomide was 24 months. The median number of administered cycles was 3; 32 of the 40 patients were alive at the time of the report. There were seven early deaths, four with Mayo stage 2 cardiac disease and three with end-stage renal disease. Three patients withdrew because of therapy-related toxicity. Grade 2 fatigue was present in 14. A worsening of renal function was seen in seven patients with preexisting renal failure (three went on dialysis), and infections of grade 2 or greater occurred in two. Three patients had a rash. There was one complete remission of the light chain disorder; 46 % had a partial hematologic response after a median of three cycles of lenalidomide–dexamethasone [146].
A multicenter phase 1/2 dose-escalation study of lenalidomide in combination with melphalan and dexamethasone has been reported. Lenalidomide was used at 15 mg per day and was escalated in cohorts to determine the maximum tolerated dose. Melphalan dosing was 0.18 mg/kg per day from days 1–4 of each 28-day cycle; dexamethasone was 40 mg on days 1–4 every 28 days. This dose of melphalan was reduced by 28 % from the standard myeloma dosing to account for the myelosuppressive effect of the combination with lenalidomide. No toxicity was reported at lenalidomide doses of 5, 10, and 15 mg. Nine additional patients are scheduled to be enrolled in a phase 2 component at 15 mg per day [147]. The Greek Myeloma Study Group reported a phase 1/2 study of lenalidomide with intermediate-dose dexamethasone and low-dose cyclophosphamide for the treatment of AL amyloidosis [148]. Dexamethasone, 20 mg, was administered on days 1–4, cyclophosphamide on days 1–10, and lenalidomide on days 1–21, in a phase 1 dose-escalation scheme in which the highest achievable dose of lenalidomide was 25 mg and that of cyclophosphamide was 100 mg. Eligibility for participation required a serum creatinine level less than 2.5 mg/dL, total bilirubin less than 0.5 mg/dL, and aspartate aminotransferase and alanine aminotransferase levels less than five times the upper limit of normal. Ten patients enrolled, 3 at dose level 0 and 7 at dose level 1, which represents lenalidomide at 10 mg per day for 21 days and cyclophosphamide at 100 mg per day for 10 days. One patient at dose level 1 had a grade 3 deep vein thrombosis. Other toxicities were hematologic. Six patients were evaluable for response; three achieved a partial remission. There was one death due to progressive amyloidosis. The maximum tolerated dose has not been reached. It appears the combination of cyclophosphamide, lenalidomide, and dexamethasone has potential to enhance the treatment of AL amyloidosis. Eight patients ineligible for the protocol because of renal failure, including three on dialysis, received cyclophosphamide, lenalidomide, and dexamethasone on a compassionate-use basis. One discontinued treatment because of toxicity. Four patients continued therapy, and one patient on dialysis achieved a complete hematologic response [148].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


