Funding . The work was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health.
Disclosure Statement: Dr. Stratakis holds patents on technologies involving PRKAR1A, PDE11A, GPR101 genes and/or their function; his laboratory has received research funding support by Pfizer Inc. for work unrelated to this project.
Introduction
Serum sodium is tightly regulated by water homeostasis, which is primarily mediated by arginine vasopressin (AVP), thirst, and the kidneys. If water balance is disrupted, abnormalities in serum sodium concentration may occur. Hypernatremia, defined as a serum sodium above 145 mmol/L (145 mEq/L), is a common electrolyte disorder. As sodium is a functionally impermeable solute and is the major solute contributing to osmolality, hypernatremia results in cellular dehydration. Hypernatremia may result from excess loss of solute-free water, decreased solute-free water intake, or excess sodium administration. Although correction of transient hypernatremia is usually well tolerated, correction of chronic hypernatremia (with resultant chronic plasma hypertonicity) should be administered slowly and cautiously to avoid serious or even life-threatening consequences. ,
Physiology of Water and Sodium Homeostasis
Serum osmolality, determined primarily by serum sodium, is tightly regulated by water homeostasis. This balance is maintained by thirst, AVP, and the kidneys. In normal humans, osmolality, or the concentration of osmotically active solutes in body fluids, is remarkably constant despite large variations in water and solute intake and excretion. Every kilogram of body water contains 285 to 290 mOsm of solute, consisting primarily of salts of sodium in extracellular fluid and of potassium in intracellular fluid. The identical osmolality of intracellular and extracellular fluids is produced by free movement of water across cellular and subcellular membranes, governed by the physical forces of osmosis and diffusion. A gain or loss of free water will be shared by all major body compartments (vascular, interstitial, and intracellular) in proportion to their relative sizes. The single exception to this free movement is the control of water permeability of the distal portion of the nephron by AVP (or antidiuretic hormone). This ability to excrete a hyperosmotic urine allows conservation of free water when the supply of available water is limited.
The day-to-day fluctuations in total body water in a healthy individual are very small, amounting to approximately 0.2% of body weight per 24 hours. Although infants have a relative excess of body water and extracellular volume as related to total body weight, the surface area, oxygen consumption, cardiac output, insensible water loss, renal water excretion, and overall metabolism are all high in relation to total body water. Therefore, infants and young children are more vulnerable to water deficit and dehydration than adults. Even with maximal renal conservation of water, the body is unable to prevent a continuous loss of insensible fluid through the skin, lungs, and gastrointestinal tract. For the replacement of this extrarenal loss, a person must rely on adequate water ingestion, with thirst playing an essential role in the regulation of tonicity and volume of body fluids.
The major stimulus to thirst is increased osmolality of body fluids as perceived by osmoreceptors in the periventricular nucleus of the hypothalamus. Hypovolemia also has an important effect on thirst which is mediated by arterial baroreceptors and by the renin-angiotensin system. Although thirst is a conscious sensation, hypothalamic stimulating and inhibitory impulses are transmitted to the cerebral cortex and consciousness, which transforms the need, or lack of need, for water into appropriate behavior. Furthermore, impulses of cortical or voluntary origin can readily condition the sensation of thirst and create what might be called the thirst appetite, or voluntary habits of drinking. These vary greatly from individual to individual, with psychogenic polydipsia representing the extreme form of excessive ingestion. Conversely, in humans, injury to these centers, or failure of these hypothalamic-cortical impulses to stimulate thirst in a patient with altered mental status, may cause inadequate water ingestion despite need.
Similar to thirst, change in body fluid osmolality is the most potent factor affecting AVP secretion. However, hypovolemia, the renin-angiotensin system, hypoxia, hypercapnia, hyperthermia, physical stress, and pain also have important effects, and many drugs have been shown to stimulate the release of AVP as well. AVP is a hormone produced by magnocellular neurons of the supraoptic and paraventricular nuclei of the hypothalamus. AVP is produced as a pre-prohormone, pre-proAVP. After cleavage of pre-proAVP, AVP is produced, along with neurophysin II and copeptin. Whereas neurophysin II functions as carrier protein for AVP, the function of copeptin has yet to be understood. AVP then travels in neurosecretory granules along the axons of the magnocellular neurons via the neurohypophyseal tract of the pituitary stalk and terminates at the posterior pituitary, where AVP is released into circulation. When serum osmolality rises above approximately 284 mOsm/kg H 2 O, AVP is released into circulation from the posterior pituitary. AVP then acts at the renal collecting ducts where it binds to its receptor, AVPR2 (or V2 receptor), which is expressed at the basolateral surface of the principal cells of the collecting ducts. Binding of AVP to its receptor results in translocation of aquaporin 2 to the apical membrane, allowing water to move from the lumen of the collecting ducts into the principal cell and then enter the interstitium through aquaporin 3 and 4 channels. The overall effect is resorption of solute-free water with concentrated urine and decreased urine output. Small changes in plasma AVP concentration of 0.5 to 4 μU/mL have major effects on urine osmolality and renal water handling.
Whereas renal water handling is primarily determined by circulating AVP and renal perfusion, reabsorption of sodium throughout the nephron is facilitated by several luminal sodium transporters along the nephron, which take advantage of low concentrations of sodium within the cells maintained by sodium/potassium-ATPase (Na/K-ATPase) on the basolateral membrane. In the proximal tubule, the majority of the sodium is reabsorbed via the sodium/hydrogen exchanger NHE3, and angiotensin II increases sodium reabsorption by NHE3. The Na/K/Cl cotransporter in the thick ascending limb of Henle is the primary transporter. In the collecting duct, aldosterone activity promotes sodium reabsorption and, as mentioned previously, vasopressin activity promotes reabsorption of water.
Etiologies of Hypernatremia
Hypernatremia may occur due to an excess loss of solute-free water, decreased solute-free water intake, or excess sodium administration ( Fig. 19.1 ). Although solute loss may occur with excess solute-free water loss, decreased solute intake may occur with decreased solute-free water intake, and solute-free water intake may occur with excess sodium administration, the balance of solute and solute-free water changes will determine the serum osmolality. This balance is also important to recognize clinically, because the balance of solute and solute-free water changes will impact the assessment of volume status during physical examination (discussed in more detail in the “Clinical Presentation of Hypernatremia” section).
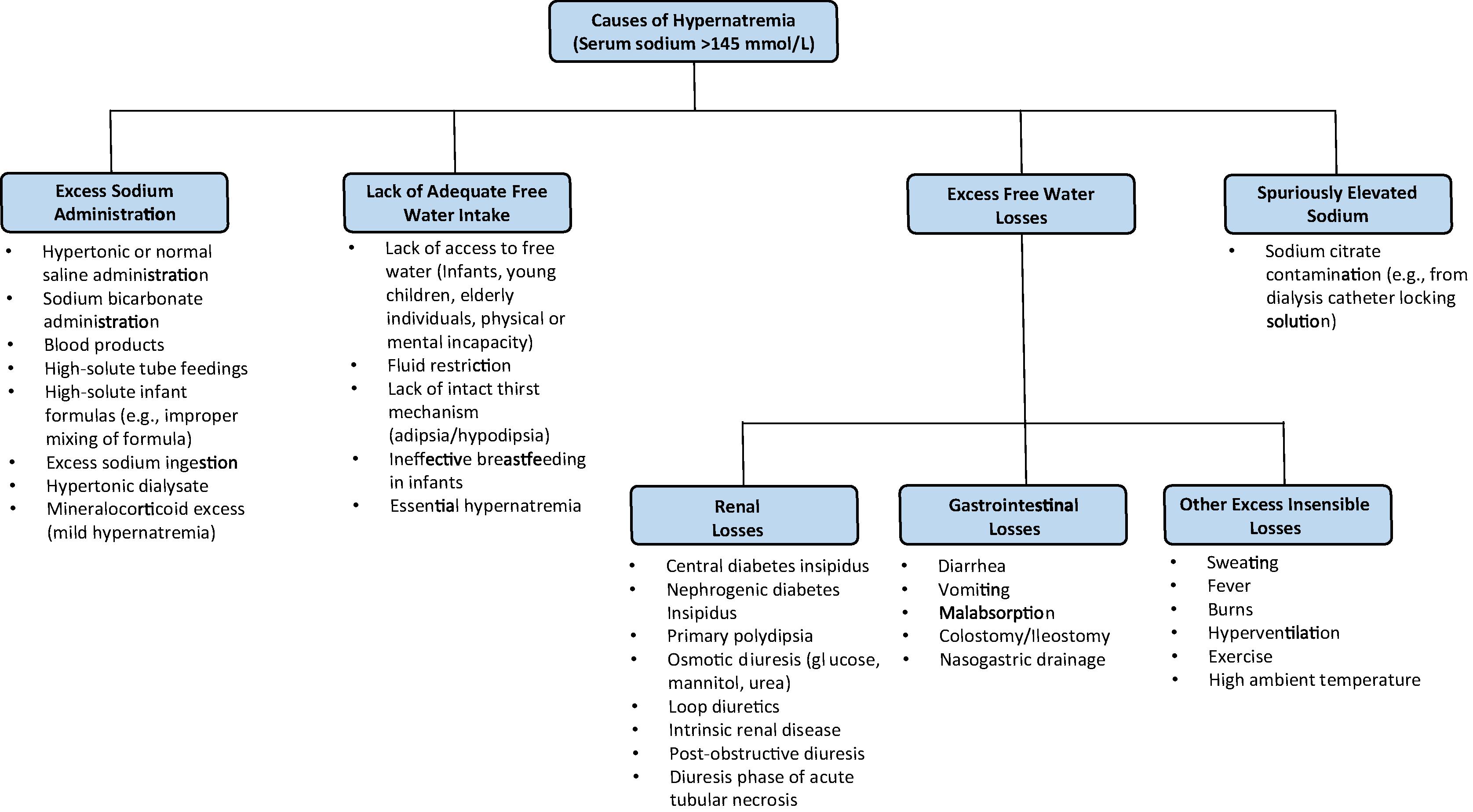
The most common cause of hypernatremia is lack of adequate free water intake. As serum sodium is tightly regulated, persistent hypernatremia should not occur in an individual with access to free water, who is alert, and has an intact thirst mechanism. Those at highest risk of hypernatremia are infants, elderly persons, those with altered mental status or neurologic impairment, or intubated patients. In these patients, although their thirst mechanism is intact, they cannot independently access fluids and are unable to communicate their need for fluids. Infants, especially those born preterm, are at particularly high risk for the development of hypernatremia because of their relatively small mass-to-surface area ratio and their dependency on a caretaker to administer fluids. Additionally, ineffective breastfeeding is a rare cause of hypernatremia in neonates, and it may be accompanied by significant vascular complications, such as venous thrombosis. , Close medical follow-up after birth with careful attention to weight loss and breastfeeding adequacy has been shown to prevent potentially devastating complications of hypernatremia in these young patients. In elderly individuals, hypernatremia is often associated with infirmity or febrile illness. Thirst impairment also occurs in elderly patients, and nursing home residents and hospitalized patients are prone to hypernatremia because they depend on others for their water requirements. Additionally, in a patient who is fluid restricted (e.g., nothing by mouth), hypernatremia may occur. Critically ill patients are also at higher risk of hypernatremia with approximately 2% to 6% hypernatremic on intensive care unit (ICU) admission and 4% to 26% becoming hypernatremic during the course of an ICU admission. Patients with a lack of intact thirst mechanism (adipsia/hypodipsia) due to hypothalamic defects or with an altered set point for thirst and AVP release (termed “essential hypernatremia”), hypernatremia may occur due to lack of adequate free water intake.
Excess free water loss is another important cause of hypernatremia and includes both renal and insensible losses of water. Renal losses of excess water include central diabetes insipidus (DI), nephrogenic DI, primary polydipsia, osmotic diuresis, diuretic use, intrinsic renal disease, postobstructive diuresis, and the diuretic phase of acute tubular necrosis. Central DI occurs due to deficient AVP synthesis or secretion, and it results in the inability to maximally concentrate urine, resulting in water diuresis. Central DI may be hereditary, such as from autosomal dominant variants in AVP or autosomal recessive variants in the AVP , WFS1 , or PCSK1 genes. In general, however, central DI is most often acquired, resulting from hypothalamic, pituitary stalk, or posterior pituitary involvement of intracranial neoplasia, infiltrative lesions, inflammatory processes, or traumatic injury; central nervous system (CNS) infections; or drug or toxin exposure. Nephrogenic DI is due to renal resistance of AVP, and it presents clinically identical to central DI with water diuresis. Nephrogenic DI may be hereditary, such as from X-linked variants in the receptor gene AVPR2 or autosomal recessive or autosomal dominant AQP2 gene variants. Acquired causes of nephrogenic DI include drug exposure (e.g., lithium, cisplatin, demeclocycline), hypokalemia, hypercalcemia, infiltrative lesions, vascular disorders (e.g., sickle cell anemia), or mechanical (e.g., polycystic kidney disease, urinary tract obstruction). Gestational DI may occur during pregnancy and is due to increased placental vasopressinase activity with increased breakdown of AVP. Primary polydipsia is a primary excess fluid intake, and it is associated in some with psychiatric conditions, though may also be seen in compulsive water drinking or due to an abnormally low thirst threshold (termed “dipsogenic DI”). Although hyponatremia may also occur in primary polydipsia, hypernatremia may occur due to tonicity of body fluids stabilizing at a new set point around the osmotic threshold for AVP secretion, due to depletion of renal medullary solute (“renal washout” effect), and due to downregulation of aquaporin 2 channels. Similar to all forms of DI, these mechanisms in primary polydipsia may prevent maximal concentration of urine with resultant water diuresis. Osmotic diuresis may also result in excess renal water losses and may be due to glucosuria (e.g., diabetes mellitus, sodium-glucose cotransporter 2 inhibitors), mannitol, or urea. Gastrointestinal losses of water are a common cause of excess water loss. Infants, young children, elderly patients, patients with DI, or other preexisting risk factors for hypernatremia are at the highest risk of severe hypernatremia from gastrointestinal losses, especially diarrhea. Other causes of excess insensible water losses include fever, hyperventilation, burns, exercise, excess sweating, and high ambient temperatures.
Hypernatremia may also occur due to excess sodium intake, such as from hypertonic or even normal saline infusions, sodium bicarbonate administration, blood products, high-solute tube feedings, or excess salt ingestion (e.g., salt tablets). High-solute infant formulas may also contribute to hypernatremia in infants, which may occur with improper mixing of formula. Mineralocorticoid excess may also result in hypernatremia, together with hypokalemia and hypertension, though hypernatremia is typically mild with serum sodium typically less than 150 mEq/L due to persistent volume expansion resulting in increasing the set point for AVP and thirst.
Clinical Presentation of Hypernatremia
The signs and symptoms of acute hypernatremia largely reflect CNS involvement and are more prominent when the increase in the serum sodium concentration is large or occurs rapidly over a period of hours. Most outpatients with hypernatremia are either very young or very old, due to the previously mentioned risk factors for decreased free water intake. Common symptoms in infants include nonspecific manifestations such as tachypnea, weakness, muscle twitching, irritability, a characteristic high-pitched cry, vomiting, insomnia, and fever. Unlike infants, adults and geriatric patients generally have fewer symptoms until serum sodium exceeds 160 mmol/L. Because the most common cause of hypernatremia is excessive solute-free water loss, patients may also have evidence of hypovolemia, including tachycardia, orthostatic blood pressure changes, or decreased blood pressure, dry mucous membranes, and delay in capillary refill. , However, volume depletion is variable, depending on the balance of water deficit and sodium excess. As water moves out of brain cells leading to contraction of cells of the brain, vascular rupture may occur in severe cases, with cerebral bleeding, subarachnoid hemorrhage, and progressive neurologic decline, including altered mental status, lethargy, coma, seizures, irreversible neurologic injury, and death. , , As can be seen in rapid correction of hyponatremia, if serum sodium rises quickly, central pontine myelinolysis may also occur in hypernatremia.
In chronic hypernatremia, defined as hypernatremia present more for than 1 day, patients may be asymptomatic due to cerebral adaption. In early adaptation (within a few hours), there is accumulation of electrolytes into cells of the brain, pulling water into the cells. In chronic adaptation (occurs in hours to days), normalization of brain volume occurs as a result of accumulation of organic osmolytes. These solutes were historically termed “idiogenic osmoles” because they were unidentified and thought to be produced by the brain cells themselves. , , Because of the adaptation that occurs, neurologic symptoms are less likely to be induced compared to acute hypernatremia. However, assessment of patients with chronic hypernatremia can be complicated by comorbid neurologic disease, as patients with an intact thirst mechanism (assuming access to free water) would have a compensatory increase in thirst preventing sustained hypernatremia, even for individuals with DI.
The concept of cerebral adaptation to hyperosmolality becomes clinically relevant regarding management of hypernatremia (discussed in detail later). Although normalization of brain volume occurs, this cerebral adaptation does not correct the hyperosmolality in the brain. Therefore, in patients with chronic hyperosmolality, aggressive treatment with hypotonic fluids may cause cerebral edema as water uptake by brain cells occurs more rapidly than osmolytes leave the brain cells, which can lead to coma, convulsions, and death. This devastating consequence has been primarily described in children and highlights the importance of slower corrections of hyperosmolality.
Diagnostic Evaluation
Hypernatremia is often multifactorial, and a thorough history is key to narrow down the differential, including a review of fluid intake to determine if the patient has an intact thirst mechanism, has restricted access to fluids, or is not being provided adequate free water in intravenous fluids. Patients should be evaluated for gastrointestinal losses, dermal losses from fever or burns, diet history (including enteral feedings, mixing of formula, breastfeeding adequacy), medication history (including diuretics), and sources of exogenous sodium. Strict intake and output should be followed to assess for polyuria (suggestive of DI, primary polydipsia, osmotic diuresis, or intrinsic renal disease) or oliguria (suggestive of hypovolemia or acute kidney injury). Serum sodium, glucose, blood urea nitrogen, creatinine, calcium, potassium, serum osmolality, urine osmolality, and urinalysis for glucose and specific gravity should be measured. Serum sodium should be measured to confirm the presence of hypernatremia and to calculate the free water deficit. Urine osmolality and urine specific gravity should be determined to assess if the renal concentrating ability is appropriate, as urine should be maximally concentrated (urine osmolality greater than 800 mOsm/kg H 2 O) in the setting of hypernatremia because hyperosmolality results in a maximal stimulus for AVP release. , ,
If the hypernatremic patient is determined to have polyuria, defined as urine output of greater than 2 L/m 2 per 24-hour (approximately 150 mL/kg per 24-hour at birth, 100 to 110 mL/kg per 24-hour until the age of 2 years, and 40 to 50 mL/kg per 24-hour in the older child and adult), then the patient should be assessed for causes of osmotic diuresis (e.g., glucosuria, uremia, mannitol use). Serum calcium and potassium should also be evaluated as hypercalcemia and hypokalemia can result in acquired nephrogenic DI. Serum and urine creatinine, as well as urine sodium, should be assessed to determine if there is renal failure or inappropriately low or normal urinary sodium.
Serum osmolality and urine osmolality should also be assessed if polyuria is present, with a high serum osmolality (greater than 300 mOsm/kg H 2 O) with an inappropriately low urine osmolality (less than 300 mOsm/kg H 2 O) being consistent with the diagnosis of DI, though it does not distinguish between nephrogenic and central DI. A low or normal serum osmolality (less than 270 mOsm/kg H 2 O), which would not be expected in the setting of hypernatremia, with an appropriately high urine osmolality (greater than 800 mOsm/kg H 2 O) may be consistent with primary polydipsia. A serum osmolality between 270 and 300 mOsm/kg H 2 O with a urine osmolality less than 800 mOsm/kg H 2 O is nondiagnostic, and an indirect water deprivation test may be indicated to differentiate between DI and primary polydipsia, though it has limited diagnostic accuracy, particularly for distinguishing primary polydipsia from partial central DI. A random plasma copeptin may be considered, with a high diagnostic accuracy of a plasma copeptin level greater than 21.4 pmol/L in identifying nephrogenic DI from central DI and primary polydipsia. , Additionally, two new promising diagnostic tests, the hypertonic saline test and arginine-stimulated copeptin test, both of which utilize stimulated plasma copeptin measurements, have been proposed as alternative diagnostic tests for differentiating central DI and primary polydipsia. , , However, these latter tests have not yet been validated in the pediatric population.
Management
The initial step in management of patients with hypernatremia is to determine the volume status. Although isotonic saline is not appropriate for correcting hypernatremia, the exception to this is in the initial management of hypovolemia and replacement with 0.9% saline to improve hemodynamic stability. However, after circulatory stabilization, fluids should then be switched to a hypotonic solution (e.g., oral free water or intravenous 0.45% saline) to continue correction of the free water deficit. The free water deficit is determined as follows:
Free Water Deficit = Body weight in kg × 0.6 × (Measured Na/Ideal Na – 1)
As 60% of body mass is water (50% in female adults and 45% to 50% elderly individuals), multiplying 0.6 times the body weight allows for calculation of total body water. Importantly, the free water deficit is an estimate of the free water deficit with its own limitations (e.g., lack of knowledge of premorbid body weight, estimation of approximately 60% of body mass being water), and serum sodium should be closely monitored during the correction to prevent too rapid a decrease in sodium. Additionally, the free water deficit does not account for ongoing urinary and insensible losses, which should be added amount of fluids to be given.
Once the free water deficit is determined, the next step is to determine the rate to replace the free water deficit. The rate at which the deficit may be safely administered depends on how quickly hypernatremia developed. If the hypernatremia developed within only a few hours, then likely only early cerebral adaptation has occurred, and with correction of hypernatremia, the brain cells can rapidly lose potassium and sodium in response to decreasing serum osmolality. Therefore, correction of hypernatremia may safely be provided more quickly than in chronic hypernatremia. An appropriate correction of acute hypernatremia would be decreasing serum sodium by 1 mEq/L per hour. However, with chronic hypernatremia, loss of accumulated organic osmolytes in brain cells occurs more slowly as hypernatremia is corrected. As fluids are given to correct the hypernatremia, more water is pulled into the brain cells still containing the organic osmolytes, which may result in cell swelling and cerebral edema, with potentially devastating neurologic consequences. , This development of cerebral edema has been primarily described in children in whom the hypernatremia was corrected at a rate exceeding 0.7 mEq/L per hour. In comparison, no neurologic sequelae were induced if the plasma sodium concentrations were lowered at 0.5 mEq/L per hour. Often it is unclear when the hypernatremia began to develop, in which case a slower correction should be given, as in chronic hypernatremia, for which serum sodium should be lowered by no more than 0.5 mEq/L per hour and no more than by 12 mEq/L per day. Additionally, the free water deficit correction does not include urine output and insensible losses, so maintenance fluids should be given in addition to the free water deficit.
The rate of fluids also depends on the choice of fluids given. If the patient is able to tolerate enteral fluids, free water given by mouth or by tube is the preferred method to correct hypernatremia. However, if the hypernatremic patient is unable to tolerate enteral fluids, then the free water deficit correction may be given with hypotonic intravenous fluids (e.g., 0.2% saline or 0.45% saline). If free water by mouth is given, then a free water deficit may be calculated that aims for a goal sodium that is 12 mEq/L or less, then dividing that rate by 24 hours, which would give a rate that is estimated to decrease serum sodium by no more 12 mEq/L in a 24-hour period. If intravenous fluids are given, then a greater volume of fluids would be needed to correct the same free water deficit for increasing concentrations of saline. For example, 0.2% sodium chloride in water is approximately 75% free water, and 0.45% sodium chloride in water is approximately 50% water. Therefore, it would require more 0.45% sodium chloride in water to correct the same free water deficit as compared with 0.2% sodium chloride in water. Importantly, serial serum sodium levels should be assessed to confirm that serum sodium is not decreasing more quickly than expected to allow titration of the free water deficit correction accordingly.
For treatment of central DI desmopressin is given, either as intranasal spray or tablets. Careful monitoring is needed to avoid the complications of water intoxication and hyponatremia, which may be prevented by allowing polyuria and thirst to “breakthrough” approximately once a day, or at least once a week at a minimum. However, in infants central DI is often managed with a low-solute infant formula and a thiazide, though polyuria will persist with this regimen, and desmopressin may become necessary. Management of nephrogenic DI includes removal of precipitating drugs (if possible) and sometimes initiation of thiazide diuretics, or nonsteroidal antiinflammatory drugs. For patients with a lack of intact thirst mechanism or for young infants, a water prescription may be needed in addition to the aforementioned management.
Prognosis
The morbidity and mortality associated with hypernatremia varies widely according to the severity of the condition, the rapidity of its onset, and the underlying condition resulting in hypernatremia. , , Most deaths seem to be independent of the severity of hypernatremia and not directly related to CNS complications. Patients who develop hypernatremia following hospital admission and patients in whom treatment is delayed have the highest mortality. , ,
References
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


