Desmoid-type fibromatosis is a rare nonmetastasizing neoplasm with variable behavior. Recent discoveries into the biology of this disease hold promise for identifying prognostic and predictive features and novel therapeutic targets. Surgery has been the historical standard of care but carries considerable drawbacks in terms of high local recurrence rates and poor functional outcomes. Improved understanding of the natural history of desmoid-type fibromatosis has resulted in a paradigm shift toward nonoperative management. Effective medical treatment options include nonsteroidal antiinflammatory drugs, hormone therapy, cytotoxic chemotherapy, and targeted agents. A treatment algorithm has been proposed with the objective of optimizing treatment.
Key points
- •
Desmoid-type fibromatosis is a clonal proliferation without metastatic potential; mutations of the CTNNB1 gene are documented in sporadic disease.
- •
Historically surgery has been the standard of care, but resultant functional impairment and high local recurrence rates have contributed to a paradigm shift toward more conservative management.
- •
Multiple pharmacologic therapies have been shown to be effective, including chemotherapy, hormone therapy, targeted therapies, and nonsteroidal anti-inflammatory drugs.
- •
Other therapeutic modalities, such as radiation therapy, cryoablation, and isolated limb perfusion can be considered in select cases.
- •
The natural history of desmoid-type fibromatosis is unpredictable and can include periods of prolonged stability or spontaneous regression.
Introduction
Definition
Desmoid-type fibromatosis (DF) is a rare benign fibroblastic/myofibroblastic proliferation that can occur in almost any anatomic location. Although the histologic appearance is consistent with fibrosis or reactive proliferation, the neoplastic nature of this disease was suggested by the discovery of a clonality in DF cells. DF lacks metastatic potential, but its propensity for local recurrence can entail considerable morbidity. In the latest edition of the World Health Organization’s classification of tumors of bone and soft tissue, DF is defined as an intermediate/locally aggressive tumor.
Introduction
Definition
Desmoid-type fibromatosis (DF) is a rare benign fibroblastic/myofibroblastic proliferation that can occur in almost any anatomic location. Although the histologic appearance is consistent with fibrosis or reactive proliferation, the neoplastic nature of this disease was suggested by the discovery of a clonality in DF cells. DF lacks metastatic potential, but its propensity for local recurrence can entail considerable morbidity. In the latest edition of the World Health Organization’s classification of tumors of bone and soft tissue, DF is defined as an intermediate/locally aggressive tumor.
Pathophysiology and emerging biological insights
Adenomatous Poliposis Coli Gene
The earliest insight into the biology of DF stemmed from the observation of a high prevalence of desmoids in patients with familial adenomatous polyposis (FAP/Gardner syndrome). FAP is driven by mutations in the Adenomatous Poliposis Coli (APC) gene and is characterized by innumerable polyps in the gastrointestinal tract (primarily the colon) that can evolve to adenocarcinoma. FAP is also associated with frequent DF, arising mainly in the abdominal cavity (mesenteric DF) or in the abdominal wall within previous surgical scars. As a general rule, FAP-related DF should be distinguished from sporadic DF, as the two exhibit different clinical behavior and necessitate different treatment strategies. APC is involved in controlling the level of β-catenin in the cytoplasm. When APC is mutated, β-catenin cannot be degraded and it translocates to the nucleus where it affects gene transcription.
β-Catenin
β-catenin is a dual-function protein encoded by the CTNNB1 gene, regulating the coordination of cell-cell adhesion in the cytoplasm as well as gene transcription in the nucleus. β-catenin is a subunit of the cadherin protein complex and acts as an intracellular signal transducer in the Wnt signaling pathway.
Generally, cytoplasmic β-catenin levels are kept in equilibrium via continuous ubiquitin-proteasome–mediated degradation, a process that is regulated by a multiprotein complex containing axin, APC, GSK3β, and CK1α. β-catenin phosphorylation allows recognition by ubiquitin ligase, which targets the protein for destruction in the proteosome. Unphosphorylated β-catenin is stable and can translocate to the nucleus to interact with the TCF family of transcription factors to activate downstream genes ( Fig. 1 ).
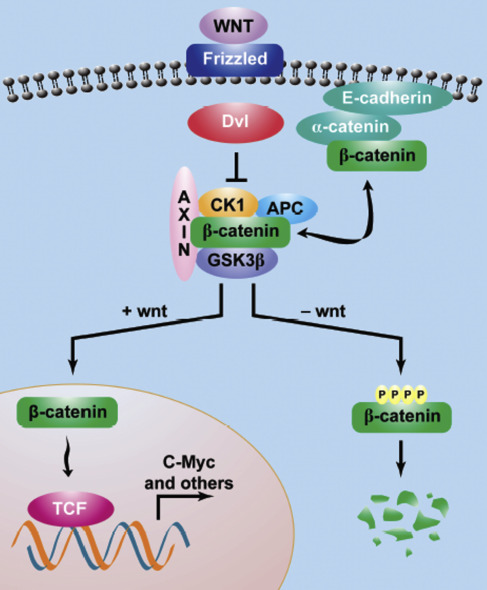
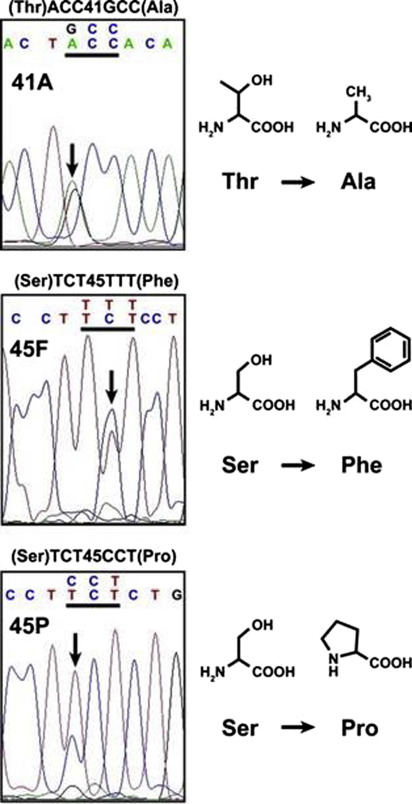
APC mutations do not in fact underlie most sporadic DF; rather CTNNB1 mutations were found in 85% to 88% of sporadic DF in historical published series. With modern genome sequencing techniques, the frequency of CTNBB1 mutations has been shown to be even higher, providing compelling evidence that alterations in the Wnt/APC/β-catenin pathway underlie the development and progression of this disease. Most of the β-catenin mutations occur in the region encoding the phosphorylation domain on exon 3. Three main point mutations have been described in codons 41 and 45, which represent targets for phosphorylation by GSK3β and CK1, respectively ( Fig. 2 ). The most common CTNNB1 mutation in sporadic DF is T41A, accounting for around 50% of cases. S45F accounts for approximately 30% and S45P for 7%. Rare mutations in exon 3, such as T40A and small in-frame deletions have been reported. Other genetic events, such as APC loss, chromosome 6 loss, or BMI I mutation, have been recently published.
Target genes of the transcriptional complex β-catenin/TCF3 in DF are largely unknown, as few studies have evaluated potential desmoid-associated gene expression deregulation. Available data on gene expression show some common deregulated genes between different published series, but the comparability of studies is limited by differences in tumor types and study technique. Despite the crucial role of β-catenin in the pathogenesis of DF, additional players must be involved as β-catenin is mutated and overexpressed in other tumors types, including hepatocellular carcinoma, colorectal, lung, breast, ovarian, and endometrial cancers.
β-Catenin Mutations and Beyond
The role of specific β-catenin mutations in DF is not yet fully understood. In surgically treated series, patients harboring a CTNNB1 S45F mutation had more aggressive disease with higher rates of local recurrence compared with CTNNB1 T41A mutants. In an international multicenter study the 3- and 5-year recurrence-free survival (RFS) rates were 49% and 45% for those with a CTNNB1 S45F mutation, 91% and 91% for wild-type (WT) genes, and 70% and 66% for all other mutations, including T41A and S45P ( P <.001). In a recent series, van Broekhoven and colleagues confirmed that an S45F mutation entailed worse RFS in patients treated surgically (5-year cumulative risk of recurrence of 63.8%); it remained an independent risk factor in multivariable analysis.
The biological effect of different β-catenin mutations is an area of ongoing active investigation, with the objective of eventually predicting disease behavior and response to treatment based on gene expression or miRNA signature. Two European trials of patients with DF being managed with a watchful-waiting strategy seek to elucidate the prognostic value of specific β-catenin mutations with respect to disease progression ( NCT02547831 ).
Hamada and colleagues showed that all patients harboring a CTNNB1 S45F mutation treated with meloxicam had progressive disease ( P = .017), whereas other mutations had no impact on response to treatment. Recently, it has been reported that specific mutations also correlate with response to imatinib in a phase II clinical trial. The progression arrest rate at 6 months was 70%, 81%, and 43% for T41A, S45F, and WT, respectively. CTNNB1 S45F mutated DF was in fact overrepresented in this trial, which included only progressing patients according to Response Evaluation Criteria in Solid Tumors (RECIST), underlying the more aggressive behavior of this specific subgroup.
Gene Profiles
The ongoing prospective European trial evaluating a watchful-waiting strategy is also focusing on the possible prognostic role of a gene signature for progression ( NCT02547831 ). A specific signature of 36 genes (including FECH, STOML2, and TRIP6) significantly associated with progression-free survival in a subgroup of patients treated with surgery will be tested in the observed population. Dufresne and colleagues previously reported the capability of a specific molecular signature consisting of 15 miRNAs to stratify time to progression following imatinib treatment.
Etiology
The etiology of DF remains unclear. DF can occur in previous surgical scars, often after caesarian section, or in the abdomen following surgical procedures, especially in patients with FAP. Although the mesentery is the preferred site for FAP-associated DF, the most common anatomic site of sporadic DF is the abdominal wall in patients with a recent history of pregnancy.
Desmoid-Type Fibromatosis and Hormones
This propensity for DF to occur following pregnancy has been postulated to reflect a degree of hormone sensitivity in the tumor within a predisposing microenvironment. However, a direct correlation between estrogen level and DF has never been demonstrated. The positive expression of the sex steroid receptors (estrogen and progesterone) was not described in all series, largely because of different antibodies used for staining. So far, no correlation between hormone receptor expression and response to specific antiestrogenic agents has been demonstrated. Moreover, the correlation between female sex, DF, and hormone therapy has never been tested in randomized controlled trials.
Desmoid-Type Fibromatosis and Scars
The occurrence of DF within or close to surgical scars is thought to reflect altered tissue repair processes, based on evidence that β-catenin plays a role in wound healing. In fact, β-catenin is transiently elevated in fibroblasts during tissue repair and forced β-catenin overexpression results in the formation of hypertrophic scars in mice. This abnormal expression is partly related to the release by platelets of growth factors and cytokines (platelet-derived growth factor [PDGF], transforming growth factor [TGF]) at the site of injury, with subsequent induction of β-catenin signaling in fibroblasts. In contrast, a decrease in β-catenin signaling is observed at late stages of the scarring process. Consistently, PDGF receptor (PDGFR) and TGF-β pathways seemed to be downregulated in DF.
In Gardner syndrome, the risk of developing mesenteric DF has been shown to be significantly lower after minimally invasive surgical procedures compared to open surgery.
Progenitor Cell
Despite DF cells morphologically resembling fibroblasts in an abundant collagen matrix, the actual progenitor cell of DF is not known. Aberrantly activated mesenchymal stromal cells (MSCs) are present in DF and genes and cell surface markers characteristic of MSCs are expressed in DF, suggesting that MSCs may be the progenitor cells of DF.
Patient evaluation
Symptoms
DF usually presents as a painless mass in the extremity, abdominal wall or trunk. Patients with intra-abdominal DF may note distension or increased abdominal girth. Rarely, DF may result in complications such as bowel perforation or obstruction ( Fig. 3 ). Pain is typically a late event, although it can occur or worsen after biopsy. Occasionally neurologic symptoms including both motor and sensory disturbances can be the direct result of DF, particularly those involving the pelvic or scapular girdle. Severe pain may require further evaluation and treatment. It is imperative to establish a relationship between the patient’s pain and the location or progression of the tumor, and to rule out other possible etiologies. Often the pain is not directly related to disease progression, though symptoms are clearly related to DF itself. In these cases, targeted intervention for the DF is not indicated, and multidisciplinary evaluation may be required for adequate pain therapy.
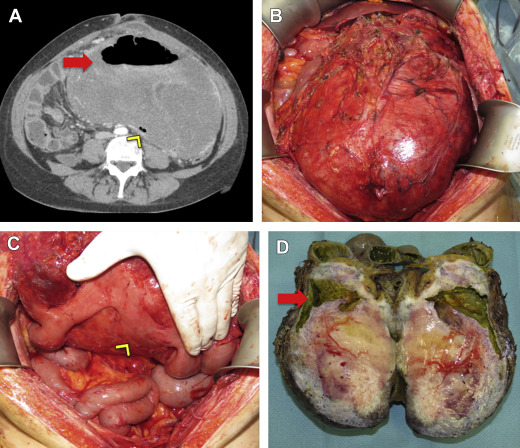
DF usually presents as a single lesion but rarely can be multifocal. Multifocality typically occurs in the extremities and in cases of recurrence after previous surgery.
Radiology
Patients with suspected DF are often first evaluated by ultrasound, although sonographic features are fairly nonspecific and further imaging is mandatory. Contrast-enhanced MRI or computed tomography (CT) is required for adequate imaging of intra-abdominal lesions. On CT, DF is isodense to skeletal muscle with inhomogeneous foci corresponding to collagenous or myxoid areas. Contrast enhancement is variable and often prominent, reflecting a rich vascularity. Contrast-enhanced CT is the preferred modality for intra-abdominal DF, as it allows delineation of the tumor from surrounding fat with less peristaltic artifact than MRI.
MRI is the imaging modality of choice for DF arising in the extremities, pelvis, perineum, pelvic and scapular girdles, superficial trunk, and head and neck region. Specific MRI features of DF have been described and have been correlated to disease status. During disease progression, DF is heterogeneously hyperintense on T2-weighted (T2W) images (corresponding to high cellularity), with avid enhancement after administration of intravenous gadolinium and bands of low signal in all sequences. In approximately 90% of cases, the key diagnostic feature of hypointense bands is identifiable on T2W images, which correspond to the dense assemblies of collagen bundles seen histologically. Spontaneous regression and dimensional response to treatment are often predicted by altered characteristics on MRI: increased T2 hypointensity and decreased enhancement ( Figs. 4 and 5 ).
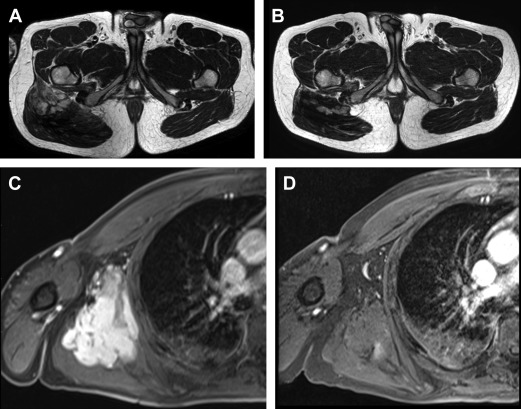
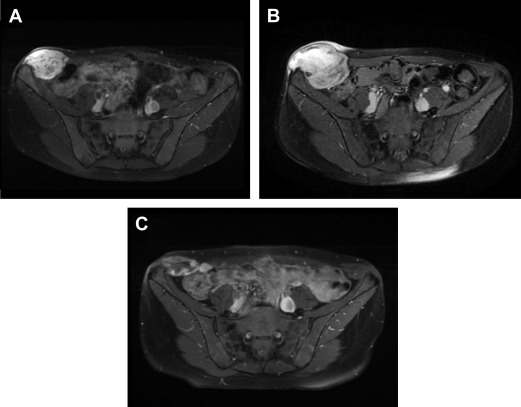
RECIST criteria for response to treatment are of little utility in evaluating DF. As these are typically slow-growing lesions, a dimensional response to treatment may not be evident for several months, despite effective therapy. Ongoing investigation into changes in both size and tumor composition on MRI are expected to assist with radiologic surveillance of DF, but at present no formal criteria exist for evaluating response to treatment.
Fluorodeoxyglucose-PET is not currently used in clinical practice, but it has been investigated in the evaluation of DF. Kasper and colleagues demonstrated a 30% decrease in the mean standardized uptake value (SUV) of serial PET scans in patients treated with imatinib. Baseline SUV values are relatively low in DF, compared with high-grade sarcomas. However, the initial mean SUV1 and maximum SUV1 (SUV1max) data suggest that there may be a role for PET in evaluating the response to therapy in DF.
Pathology
DF is composed of proliferations of spindle cells resembling fibroblasts with no malignant features (eg. hyperchromatic nuclei, atypia). These cells are embedded in a dense collagen matrix with variably prominent blood vessels and tend to have an infiltrative growth pattern. Immunostaining is positive for β-catenin (predominantly nuclear) and vimentin, but it should be noted that nuclear staining for β-catenin is not specific for DF. Other mesenchymal neoplasms, such as solitary fibrous tumor, endometrial stromal sarcoma, and synovial sarcoma can also stain positive for nuclear β-catenin. DF is negative for smooth muscle actin, desmin, h-caldesmon, CD34, c-KIT (CD117), and S-100 and positive for cyclooxygenase 2 (COX-2) and the tyrosine kinase receptor PDGFRb. It is frequently positive for androgen receptor and estrogen receptor beta but not for estrogen receptor alpha.
Given the high incidence of CTNNB1 mutations in sporadic DF, mutational analysis has become a useful diagnostic tool in challenging cases. For instance, in patients with suspected recurrent DF or patients operated on for different diagnoses (eg. retroperitoneal dedifferentiated liposarcoma) who develop new lesions in the operative field, CTNNB1 mutational analysis may be essential for diagnosis. Point mutations in exon 3 are pathognomonic of DF among spindle cell lesions. They do not occur among other soft tissue sarcomas, gastrointestinal stromal tumor, or reactive processes (eg. nodular fasciitis, myositis, and scars). However, CTNNB1 mutations are reported in several epithelial neoplasms.
There are insufficient data at present to use CTNNB1 mutation status to predict response to treatment, although a correlation has been identified in surgically treated patients. Data from the ongoing prospective trial on surveillance of primary DF may eventually add new insight in this area.
Evolving role of surgery
Surgical Outcomes in Historical Series
Prior to the year 2000, the management of DF mirrored that of soft tissue sarcoma, with surgery as the standard of care. Multiple retrospective single-institution case series have reported local control of DF after complete surgical resection to be approximately 80% at 5 years. Tumor location was found to be a risk factor for recurrence, with extremity DF portending a worse prognosis than abdominal wall and other sites. Recurrent disease was also a risk factor for further recurrence. Surgical margins, however, do not consistently correlate with recurrence ( Table 1 ). Of note, microscopically negative surgical margins are more difficult to obtain in DF compared with soft tissue sarcoma because of the infiltrative growth pattern of this disease. This point should be taken into account when considering surgical intervention with the possibility of resultant functional impairment.
| Author, Year | No. of Patients | Primary/Recurrence | Median Follow-up (mo) | 5-y DFS (%) | Surgical Margins as Prognostic Factor | |
|---|---|---|---|---|---|---|
| Negative Margins | Positive Margins | |||||
| Posner et al, 1989 | 128 | 78/53 | 88 | 85 | 50 | Yes |
| Ballo et al, 1999 | 189 | 85/104 | 112 | 75 | 50 | Yes |
| Merchant et al, 1999 | 105 | All primary | 49 | 70 | 78 | No |
| Gronchi et al, 2003 | 128 | Primary | 130 | 82 | 79 | No |
| 75 | Recurrence | 153 | 65 | 47 | No | |
| Huang et al, 2009 | 151 | 113/38 | 102 | 80 | 80 | Yes |
| Mullen et al, 2012 | 177 | 133/44 | 40 | 82 | 52 | Yes |
| Peng et al, 2012 | 141 | na | 26 | 53 | 53 | Yes |
| Crago et al, 2013 | 439 | 382/113 | 60 | 69 | 69 | No |
| Zeng et al, 2014 | 233 | 156/77 | 54 | 74 | 74 | Yes |
| He et al, 2015 | 114 | 79/35 | 73 | 85 | 50 | Yes |
Postsurgical Nomogram
A recently published nomogram incorporates tumor site, size, and patient age in estimating risk of local recurrence in patients undergoing surgical resection. As described earlier, abdominal wall DF entails a favorable prognosis, whereas extremity DF portends the worst prognosis. Increased tumor size is associated with continuously increasing risk of local recurrence, and younger patients have the highest risk of recurrent disease.
Nonoperative Management
In recent years, evidence of the unpredictable behavior of DF, including long-term disease stabilization and spontaneous regression, has resulted in a paradigm shift toward more conservative treatments.
In 1999, Lewis and colleagues adopted a watchful waiting approach to multiply recurrent DF on the basis that further surgery would result in considerable functional impairment, and demonstrated some long-term disease stabilization without further intervention. Taking into consideration these results along with the local recurrence rate of 20% to 30% (exceedingly high for a benign condition) and the potential morbidity of resection, a nonoperative approach to primary DF was advocated as first-line therapy. This approach initially entailed liberal use of medical and, to a lesser extent, radiation therapy, but eventually a period of observation with no active treatment was systematically offered to most patients with a new diagnosis of primary DF.
In the last 8 years, multiple series of a nonsurgical or observational approach to DF have been published ( Table 2 ), with the following major findings:
- •
Nonoperative management of DF can be safely offered to most patients.
- •
Disease stabilization is documented for roughly 50% of patients and for a median duration of 14 to 19 months.
- •
Only a minority of patients require surgical resection after initial observation (16% of abdominal wall DF, 4% of extra-abdominal DF).
- •
Spontaneous regression is consistently reported in 20% to 28% of cases.
- •
Disease progression is exceptionally rare after 3 years of initial observation.
Related posts:
 Contemporary Management and Controversies of Sarcoma
Refinements in Sarcoma Classification in the Current 2013 World Health Organization Classification of Tumours of Soft Tissue and Bone
Contemporary Management and Controversies of Sarcoma
Refinements in Sarcoma Classification in the Current 2013 World Health Organization Classification of Tumours of Soft Tissue and Bone
 Sarcomas 2016
Sarcomas of the Breast with a Spotlight on Angiosarcoma and Cystosarcoma Phyllodes
Management of Sarcoma Metastases to the Lung
Myxofibrosarcoma
Sarcomas 2016
Sarcomas of the Breast with a Spotlight on Angiosarcoma and Cystosarcoma Phyllodes
Management of Sarcoma Metastases to the Lung
Myxofibrosarcoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


