Desmoid tumors are rare, clonal collections of benign fibrous tissue that exhibit a highly variable clinical course. This article presents a comprehensive review of desmoid tumors and summarizes the current literature pertaining to clinical presentation, diagnostic modalities, pathogenesis, prognostic factors, and management options.
Key points
- •
Desmoid tumors are rare tumors of mesenchymal origin that vary widely in presentation and behavior.
- •
Desmoid tumors can occur in association with familial adenomatous polyposis, trauma, prior surgery, or pregnancy.
- •
Sporadic desmoid tumors can be associated with somatic mutations in the β-catenin gene.
- •
Many treatment modalities have shown benefit ranging from conservative, nonsurgical approaches to aggressive cytotoxic chemotherapy.
- •
Ongoing studies on the biology of desmoid tumors are leading to more potential rational therapeutic strategies.
Introduction
Desmoid tumors, also known as aggressive fibromatosis, were first coined in the 1830s after the Greek word desmos meaning “tendon-like.” These rare tumors arise from mesenchymal cells, similar to their malignant counterpart, sarcomas. Unlike sarcomas, there is no metastatic potential for desmoid tumors. However, despite their benign classification, desmoid tumors can be multifocal, and locally infiltrate surrounding structures such that they can be a cause of both significant morbidity and, rarely, mortality.
Desmoid tumors vary widely in presentation and behavior. These collections of fibrous tissue range from being relatively indolent and asymptomatic to creating severe local symptoms with significant morbidity. Accordingly, treatments range in aggressiveness and include observation, surgery, radiation therapy, nonsteroidal anti-inflammatory drugs (NSAIDs), hormonal agents, tyrosine kinase inhibitors (TKIs), and cytotoxic chemotherapy. New molecular insights into desmoid tumors suggest potential therapeutic targets in an attempt to expand the arsenal of therapeutic options.
Introduction
Desmoid tumors, also known as aggressive fibromatosis, were first coined in the 1830s after the Greek word desmos meaning “tendon-like.” These rare tumors arise from mesenchymal cells, similar to their malignant counterpart, sarcomas. Unlike sarcomas, there is no metastatic potential for desmoid tumors. However, despite their benign classification, desmoid tumors can be multifocal, and locally infiltrate surrounding structures such that they can be a cause of both significant morbidity and, rarely, mortality.
Desmoid tumors vary widely in presentation and behavior. These collections of fibrous tissue range from being relatively indolent and asymptomatic to creating severe local symptoms with significant morbidity. Accordingly, treatments range in aggressiveness and include observation, surgery, radiation therapy, nonsteroidal anti-inflammatory drugs (NSAIDs), hormonal agents, tyrosine kinase inhibitors (TKIs), and cytotoxic chemotherapy. New molecular insights into desmoid tumors suggest potential therapeutic targets in an attempt to expand the arsenal of therapeutic options.
Epidemiology
Desmoid tumors are uncommon, with an estimated incidence of 2.4 to 4.3 per million per year, accounting for less than 3% of soft-tissue lesions. Although there is some variability, there is a 2- to 3.5-fold increased incidence in women. A wide range of ages is affected, with most cases occurring between the ages of 15 and 60 years with an average age of 36.7 years. The majority of cases are sporadic with no known predisposing factors. However, a sizeable minority of desmoid tumors occur as a consequence of the genetic syndrome familial adenomatous polyposis (FAP) or in association with pregnancy or trauma (see the section on predisposing factors).
Pathogenesis
The Wnt/β-catenin pathway drives the pathogenesis of both sporadic and FAP-associated desmoid tumors. FAP-associated tumors frequently have adenomatous polyposis coli gene ( APC ) mutations at or beyond 3′ of codon 1444. One function of APC is to regulate the protein level of β-catenin. When β-catenin is present in high concentration it binds to APC, followed by binding of the serine-threonine kinase GSK3β. This binding eventually leads to phosphorylation of sites on APC and β-catenin degradation. In cases of mutated APC a truncated APC protein is created, which is unable to degrade β-catenin appropriately. This process results in accumulation of β-catenin and its target genes, which are implicated in loss of proliferation regulation.
Approximately 85% to 90% of sporadic desmoid tumors are associated with somatic mutations in the β-catenin gene, CTNNB1 . In a study of 254 cases of sporadic desmoids, 88% had CTNNB1 mutations identified by direct sequencing, compared with no mutations detected in the control of 175 other spindle-cell lesions. These gene mutations have been found in codons 41 and 45 of exon 3 of CTNNB1 , which produce a stabilized β-catenin protein product leading to an accumulation of β-catenin in the cell.
In endothelial cells β-catenin is not only a cell-adhesion molecule, but also plays a role in nuclear transcription. Accumulation of β-catenin, by constitutive activation of the Wnt ligand pathway, loss of APC protein function, and inability to phosphorylate β-catenin or mutations in the CTNNB1 gene, allows translocation of cytoplasmic β-catenin into the nucleus and, in conjunction with other proteins, promotes abnormal proliferation.
Recognizing the central role of β-catenin in desmoid tumors, the current value of CTNNB1 evaluation in an individual patient is unclear. Some advocate that pediatric desmoid patients with β-catenin accumulation in the nucleus be evaluated for the presence of CTNNB1 mutation. If negative, APC mutation analysis should be considered, as this could potentially be the initial manifestation of FAP. Evaluation of CTNNB1 could also be used as a potential diagnostic test in tumors that are challenging to identify. In addition, targeting the Wnt/β-catenin pathway may be a future therapeutic option of value.
Clinical presentation and behavior
Desmoid tumors arise from a variety of connective tissues, including muscle, fascia, and aponeurosis, and can present at any site in the body, with extremities/trunk, abdominal wall, and intra-abdominal locations most commonly described. There is no metastatic potential for these tumors; however, they can be multifocal and locally infiltrate surrounding structures, which may be due to their lack of a pseudocapsule and tendency to spread along fascial planes and muscle.
Intra-abdominal tumors, occurring in the bowel, mesentery, or abdominal wall, are often associated with Gardner syndrome or FAP. The presentation can range from mass effect to more serious complications of mucosal ischemia, ulceration, and bleeding. In any patient presenting with an abdominal desmoid tumor, a thorough family history and workup for FAP with colonoscopy should be considered.
Extra-abdominal tumors are typically sporadic non–FAP-associated lesions. Local invasion from these tumors can cause pain, weakness, paresthesias, and neuropathies, which can lead to debilitating symptoms.
Diagnosis
Radiology
Because of the invasive nature of desmoid tumors, identifying the extent of tumor invasion onto vital structures is paramount for consideration of local therapies. Standard radiographs can provide limited information such as tumor size and location with findings that are generally nonspecific, poorly defined, and (rarely) with calcifications. Underlying bone involvement may manifest as bone erosion or cortical scalloping.
Computed tomography (CT) scans provide additional anatomic information; however, owing to variable muscle infiltration, margins or borders may be poorly identified ( Fig. 1 ). Lesions appear as either hyperdense and/or hypodense to skeletal muscle, and enhancement with contrast is not consistent. In a study comparing CT appearance with histiologic appearance of tumors, no relationship was found that could account for the variability seen on imaging.
Magnetic resonance imaging (MRI) has been proved to be superior to other imaging modalities when characterizing desmoid tumors. MRI is able to show tumor infiltration into muscle and to distinguish boundaries between vital structures and fascial planes. Central necrosis is not seen, and because of the variable cellular and collagen content of these tumors they are heterogeneous, with bands of low signal seen on standard sequencing. T1-weighted images reveal hypointense or isointense lesions, whereas T2-weighted images show mixed hyperintense lesions. Administration of intravenous gadolinium may lead to moderate or avid enhancement of the lesions; however, this is variable owing to the varying amount of collagen deposition in a tumor. MRI provides information on tumor invasion, depth, and neurovascular encasement, which is necessary when determining the feasibility of surgery ( Fig. 2 ).
Pathology
Histologically, desmoid tumors are composed of monoclonal fibroblasts appearing as spindle-shaped cells separated by an abundant collagenous matrix ( Fig. 3 ). Few atypical, pleomorphic, or hyperchromic cells are seen, and the interlaced bundles of collagen and cells form dense tumors that are without pseudoencapsulation. The infiltrating nature of these tumors is seen at muscle borders, where the tumors extend into muscle fascicles.
Immunohistochemically these tumors show positivity for β-catenin and are negative for desmin, CD34, c-kit, and S-100. In addition, desmoids can express cyclooxygenase-2 (COX-2) and estrogen-β receptors. The proliferation index, Ki-67, is typically low. Cytogenetic analyses in sporadic tumors can reveal trisomy 8 and trisomy 20, which can occur individually or be present together.
Predisposing factors
Familial Adenomatous Polyposis/Gardner Syndrome
FAP is associated with an autosomal dominant germline mutation in the APC gene. This mutation is associated with the development of hundreds of colon polyps, typically resulting in malignant transformation, osteomas, epidermoid cysts, and other soft-tissue and benign tumors. Of patients with FAP, approximately 10% develop desmoid tumors. As prophylactic treatment, a majority of FAP patients undergo colectomy for prevention of the future development of colon carcinoma. In these patients, desmoid tumors are a significant cause of morbidity and mortality because of their predilection for prior surgical sites, and on average 77% of desmoid tumors occur 5 years after colectomy. In one series, 50% of patients with FAP and prior abdominal surgery subsequently developed a desmoid tumor at surgical sites. The median age at diagnosis is 31 years, with a 1:1 male/female ratio.
In a retrospective review of registry data, 387 patients with FAP-associated desmoid tumors were identified, and within this population 53% were found to have intra-abdominal tumors, 24% abdominal wall tumors, and 9% extremity tumors. Similar to prior publications, this study reported that family history, previous abdominal surgery, and specific APC mutations (3′ of codon 1444) were risk factors associated with the development of desmoid tumors in patients with FAP.
Pregnancy
Desmoid tumors have been associated with elevated estrogen states, and have been well described as occurring in the abdominal wall during pregnancy or in the postpartum period. The connection between estrogen levels and desmoids is based on case reports and anecdotal information, and estrogen receptors are not universally found in desmoids tumors, suggesting alternative mechanisms of growth. Surgical trauma is also known to be associated with desmoid tumors, including prior cesarean sections, and this may add to the development of desmoid tumors during pregnancy. Women who develop pregnancy-related desmoids are not at increased risk for recurrent or new tumors with subsequent pregnancies.
Trauma and Prior Surgery
Surgical trauma, particularly abdominal surgeries, has been linked to the development of desmoid tumors in prior surgical areas. This phenomenon is noted in patients with FAP, pregnancy, and sporadic tumors, with one series reporting up to 28% of patients with prior surgical or penetrating trauma sites developing a desmoid tumor. The etiology of these findings has been connected to mesenchymal stromal cells and dysregulation of β-catenin either through the APC gene or CTNNB1 gene mutations, as well as to an abnormal increase in cytokines that are seen in wound healing leading to tumor growth.
Prognostic factors
The rarity of this disease, small cohorts of patients studied, and lack of randomized clinical trials leads to difficulties and variability in the identification of prognostic factors. In several series, age has been described as a prognostic factor, but results are inconsistent. Whereas some series report older age as a poor prognostic factor, others indicate younger age, and yet others did not identify age as having any prognostic significance.
Recurrences are seen in 20% to 68% of patients, typically occurring within the first 1.5 to 5 years after treatment. It is unclear as to what extent margin status after surgical excision contributes to local recurrence. Many studies cite positive margins as a negative prognostic factor, whereas other series report no significant difference in relapse with positive or negative margins. Unlike age and margin status, tumor location is more widely identified as affecting prognosis, with extra-abdominal tumors or limb and girdle tumors leading to increased risk of desmoid tumor recurrence after excision.
In a study by Salas and colleagues, 426 patients with sporadic aggressive fibromatosis were studied to determine desmoid tumor–related prognostic factors. Multivariate analysis identified age younger than 37 years, tumor size greater than 7 cm, and tumor location as factors for poor prognosis, and patients with all 3 factors had a significantly lower probability of progression-free survival.
Management
Desmoid tumors can be locally aggressive, leading to significant morbidity. Therapeutic management options are variable and include observation, surgery, radiation therapy, hormonal therapy, targeted therapy, and cytotoxic therapy. The overall survival rate is extremely good for these patients, with one study reporting a 96% 15-year survival. However, the overall survival does not reflect the morbidity of this disease on afflicted patients, who are often young and otherwise healthy. Treatment approaches for patients require individualization and a multidisciplinary team approach.
Observation
Given the highly variable disease course, the wait-and-see approach has been advocated in select populations. Prolonged stable disease and, less commonly, primary tumor regression has been reported, often in patients with recurrent disease after surgery. In a study of 142 patients with desmoid tumors treated without surgery or radiation, there was no significant difference in 5-year event-free survival (EFS) in patients treated with the wait-and-see approach in comparison with medical therapy (49.9% vs 58.6%, respectively; P = .3196), thus supporting observation as a more viable approach than systemic therapy. Using a slightly different approach, a retrospective study by Bonvalot and colleagues reported a 3-year EFS of 65% and 68% in patients with extra-abdominal desmoid tumors who were treated with R0 surgical resection in comparison with patients who were treated without surgery (observation or medical therapy). Based on these and other experiences, observation has become a very reasonable option for well-selected patients with desmoid tumors. Likewise, the latest consensus guidelines from the National Comprehensive Cancer Network (NCCN) recommends observation as a primary treatment option for surgically unresectable tumors or resectable tumors that are not symptomatic, life threatening, or causing significant impairment.
Local Treatments
Local therapies such as surgery and radiation therapy are effective modalities for the treatment of desmoid tumors. Other emerging treatments, such as cryoablation, have shown potential improvement in small to medium-sized extra-abdominal desmoid tumors.
Surgery
Surgical wide-margin resection is historically the frontline management for desmoid tumors. However, recurrence rates after surgery for cases of either positive or negative margins are variable, ranging up to 40% in some series. The infiltrative nature and tendency to invade neurovascular structures can lead to difficulties in obtaining wide-margin resections when the goal of treatment is to preserve function, and the significance of margin status is unclear, as discussed earlier. It is reasonable to consider surgical resection as dependent on the expectant surgical functional outcome when these tumors cause impairment and excess morbidity. Specifically, NCCN consensus recommendations advocate that surgical resection with either positive or negative margins is acceptable in these situations.
Radiation therapy
Radiation therapy (RT) in patients with desmoid tumors has been used in conjunction with surgery and as primary treatment in patients with unresectable tumors or in those patients for whom resection would significantly limit function. The role of adjuvant RT is unclear because there is no consensus on whether positive surgical margins are of prognostic significance, making it difficult to determine the benefit of RT after surgery.
In a review of 22 articles from 1983 to 1998, Nuyttens and colleagues found that local tumor control was significantly improved with the addition of RT after surgery or with RT alone. In patients who underwent surgery, the addition of RT to those with positive margins improved local control rates from 41% to 75%. Later reports also confirmed the benefit of adjuvant RT for local disease control and in those with positive surgical margins.
By contrast, a retrospective analysis of 104 patients who underwent surgery, radiation, or surgery plus radiation demonstrated that all 3 modalities had similar local control rates 3 years after treatment. Although there is no consensus regarding the benefit of RT used alone or in the adjuvant setting for local control, current studies report delivering a total of 50 to 60 Gy when administering radiation treatments, noting that increased doses cause significantly more complications.
As patients with desmoid tumors are often younger and have a very good long-term survival rate, the complications of RT require consideration. The most frequently reported radiation-related effects include pathologic fractures, range-of-motion limitations, limb contractures, pain, and in-field skin cancer.
Systemic Therapies
Anti-inflammatory medications
NSAIDs were first noted to be active in desmoid tumors in 1977. Desmoid tumors have been shown to overexpress cyclooxygenase, specifically COX-2, and inhibition with NSAIDs results in decreased proliferation of desmoid tumor cells. Many small case series have demonstrated the utility of NSAIDs, including sulindac and indomethacin, both alone and in combination with other agents. In a series by Tsukada and colleagues, 12 of 14 patients had a complete response, partial response, or stable disease while being treated with a median dose of sulindac, 300 mg daily. In another report by Klein and colleagues, 3 patients with FAP were treated with indomethacin, 100 mg daily; 1 patient demonstrated a complete response whereas the 2 remaining patients had progressive disease ( Table 1 ).
| Therapy | No. of Patients | Outcome | Authors, Ref. Year |
|---|---|---|---|
| Sulindac | 14 | CR/PR: 57%; SD: 29%; PD: 14% | Tsukada et al, 1992 |
| Tamoxifen or toremifene | 20 | Overall 65% response rate | Brooks et al, 1992 |
| Toremifene | 27 | PR: 22.2%; SD: 70.3%; PD: 7.4% | Fiore et al, 2011 |
| Tamoxifen or raloxifen + sulindac | 25 (17 FAP associated, 8 sporadic) | FAP: PR/CR: 29.4%; SD: 35.2%; PD: 23.5%; lost to follow-up: 11.7% Sporadic: CR: 12.5%; SD: 62.5%; PD: 12.5%; Remained in CR: 12.5% | Hansmann et al, 2004 |
Although NSAIDs have been shown to have activity in desmoid tumors, reports incorporate both sporadic and FAP-associated tumors, assess small sample sizes, and do not provide comparisons between agents, all of which create limitations in the interpretation of these data.
Hormonal therapy
Antiestrogen treatments for desmoid tumors have been described as effective in many case reports, although it is unclear as to why this strategy is useful. In 1986, Lim and colleagues analyzed 15 desmoid tumors, and found that 33% expressed estrogen receptors (ER) and 79% expressed antiestrogen binding sites (AEBS). In another study evaluating 40 desmoid tumors, all tumors specifically expressed ER-β, albeit to varying degrees. Eighty-three percent of tumors had a greater than 50% ER-β expression, and all samples were ER-α negative. In a subsequent study evaluating 59 desmoid cases, 89% were found to express ER-β and, similarly, all samples were ER-α negative. However, correlation of ER status and response to antiestrogen therapy has yet to be reported.
Although the exact mechanism of action is not known, activity of a variety of antiestrogen therapies has been reported, including tamoxifen, toremifine, progesterone, goserelin, medroxyprogesterone acetate, testolactone, and prednisolone. The largest published case series with antiestrogens was conducted by Brooks and colleagues, who reported an overall 65% rate of stabilization of disease, partial response, or complete response to toremifene or tamoxifen (see Table 1 ). More recently toremifene was studied, and the data presented in abstract form indicated that 92.5% of patients had a partial response or stable disease, with an overall 81.5% of patients showing symptomatic improvement or radiographic response. Many case reports have been published on the benefit of tamoxifen for these tumors.
Given their relatively low toxicity profile independently, combination therapy of NSAIDs plus hormonal agents has been used with possible increased efficacy. In a series of 25 patients who received sulindac and tamoxifen, approximately 75% of sporadic tumors and 65% of FAP-associated tumors had benefit that ranged from stable disease to a complete response (see Table 1 ). Major side effects included an increased incidence of ovarian cysts, chronic fatigue, and weight gain. Although response rates are relatively high, it can take many months of treatment before benefit is seen, and available data are largely retrospective. Treatment with NSAIDs and/or hormonal agents should be considered in patients with tumors that are not life threatening or causing major functional limitations.
Tyrosine kinase inhibitors
For more than 10 years, the use of TKIs has been reported in the treatment of desmoid tumors, with success. Imatinib, a selective TKI, inhibits several class-3 tyrosine kinase receptors, including abl, PDGFR, and c-KIT. The exact target of TKIs in desmoid tumors is not known, and response does not seem to correlate with c-KIT, PDGFR-α, PDGFR-β, CTNNB1, or APC mutations. Other studies also demonstrated that the efficacy is unrelated to AKT, phosphatase and tension homologue, and C-Src activity.
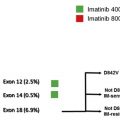
Earlier studies indicated an approximate 15% response rate to imatinib, but subsequent phase II trials favored stability of disease without robust tumor regression. The Sarcoma Alliance for Research through Collaboration conducted a phase II trial of imatinib in 51 patients with desmoid tumors, and demonstrated an overall response rate of 6% and 2-, 4-, and 12-month progression-free survival rates of 94%, 88%, and 66%, respectively. Similar findings were reported in a phase II French Sarcoma Group trial, where 40 patients with progressive disease were treated with imatinib at 400 mg daily for 1 year and then increased to 800 mg for 8 additional months. The 3-, 6-, and 12-month progression-free survival was 91%, 80%, and 67%, respectively ( Table 2 ).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


