Fig. 11.1
The sequence of events during puberty in girls. Breast bud appearance is usually before pubic hair growth; in the meantime growth velocity increases reaching the peak at Stage 4 of puberty. At this time menarche may appear [4]
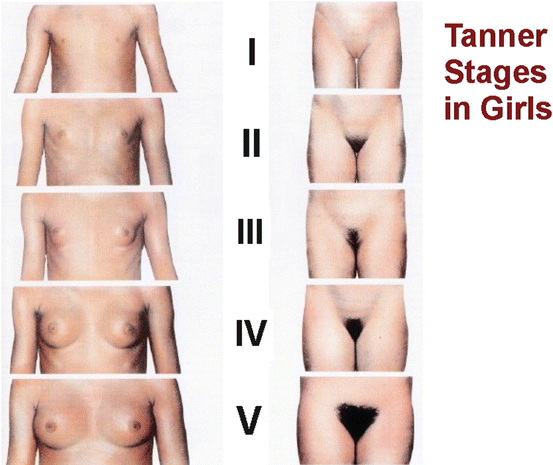
Fig. 11.2
Breast development in girls. The mammary gland grows from a breast bud that can be palpated under the nipple (Tanner stage B2) to a fully developed female breast (Tanner stage B4 or B5) over a period of 3.6 years, on average [4]
In girls, pubertal growth spurt occurs during Tanner stages 2 and 3. In girls, it occurs 2 years earlier than in boys. Girls do not show the same slowing down of growth velocity as boys before puberty and increase their growth velocity to 6 cm/year during the first year of puberty, and to 8 cm/year on average during the second year [6].
Menarche
A variety of environmental and genetic factors are involved in the regulation of menarche. The first menstrual period (menarche) occurs at an average age of 13.4 years, according to the longitudinal data obtained by Largo et al. [4]. Menarche occurs generally at Tanner stage 4. The mean age at menarche is highly correlated within families, between monozygotic twins, and within ethnic groups [7]. Twin analyses suggest that 53–74 % of the variation in age of menarche may be attributed to genetic effects [7].
11.2.2 Endocrinological Aspects of Normal Puberty
11.2.2.1 Adrenarche
Endocrinologically, the first signal for puberty is given by the adrenals (adrenarche). The onset of DHEA-S production from the adrenal zona reticularis leads to the phenomenon of adrenarche.
During infancy and early childhood, adrenal androgens (androstenedione, dehydroepiandrosterone, and dehydroepiandrosterone-sulphate) are secreted in small amounts. Their secretion increases gradually with age. This increase in androgen levels is responsible for the appearance of body odour, pubic hair and axillary hair. Therefore, pubic hair develops independently of the activation of the hypothalamic-pituitary-gonadal pathways.
Adrenarche is marked by the growth of the zona reticularis [8] and a parallel increase in the adrenal androgen levels. This phenomenon is only seen in the human beings and in some old world primates, such as the chimpanzee [9]. Plasma concentrations of the adrenal androgens increase, whereas those of cortisol remain stable, suggesting that factors other than corticotropin are involved. Hormones postulated for this role are the yet undefined androgen-stimulating factor, Corticotrophin Releasing Hormone (CRH) and more recently hormones related to body mass, such as insulin and leptin [10–13]. Although the temporal relation between adrenarche and the onset of puberty suggests that adrenal androgens might have a regulatory influence on the timing of puberty, it is now accepted that the two events are independent processes.
11.2.2.2 Regulation of the Hypothalamo-Hypophyseal-Gonadal Axis
Gonadotropin releasing–hormone
Gonadotropin releasing-hormone (GnRH), a decapeptide secreted by approximately 1000 neurons located in the basal forebrain and extending from the olfactory bulbs to the mediobasal hypothalamus, is responsible for the gonadotropin secretion by the pituitary gland. GnRH stimulates the release of LH and FSH from the pituitary which in turn stimulate the gonads. LH and FSH have negative feedback effects on the hypothalamus, whereas testosterone (T) and Androstenedione (A) produced by the testis, and Estradiol (E2) produced by the ovary, inhibit both the hypothalamus and the pituitary gland. Inhibin, activin, and follistatin have also feedback effects at both levels. GnRH secretion by the hypothalamus is under the control of a great amount of central and peripheral signals: excitatory amino acids and other neurotransmitters such GABA, gonadal sex steroids, adrenal and thyroid hormones, the GH-IGF-IGFBP axis, nutrition and related hormones such as leptin and insulin (Fig. 11.3).
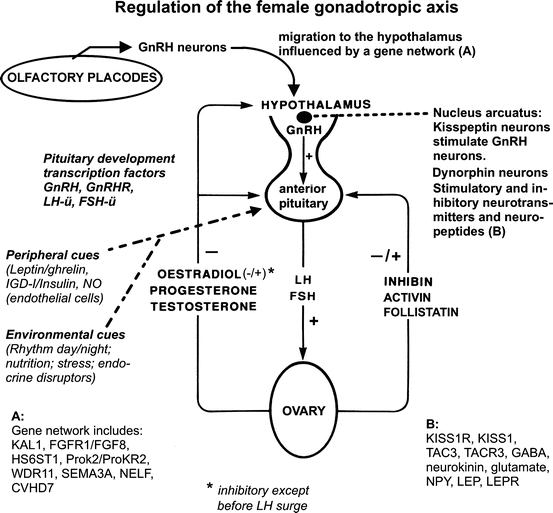
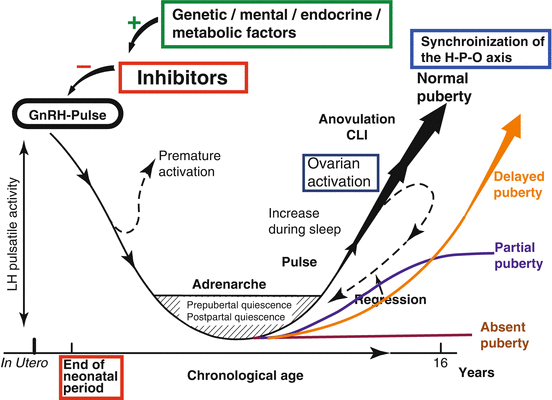
Fig. 11.3
Principals of the development and the regulation of the female gonadotropic axis (see text)
Fig. 11.4
Normal and delayed maturation of the hypothalamo-hypophyseal-gonadal axis (H-P-O-axis). Onset of puberty implies the regression of the inhibitory factors blocking the gonadal axis in childhood. Maturation and activation of the ovarian axis can be interrupted at each stage and may even regress to prepubertal quiescence
Two types of GnRH neurons have been identified to date, GnRH neuron I and II. GnRH neurons II have no known function in humans and are not involved in reproductive function, as inferred from Kallmann’s syndrome patients in whom GnRH neurons I only are affected. GnRH neurons I originate in the embryonic period and exhibit an endogenous secretion very early in development. After birth, their activity is “turned-off” by the low circulating levels of androgens/oestrogens released by the gonads, by means of a negative feedback mechanism. At puberty, the reactivation of this “gonadostat” is independent of the effect exerted by the steroids and is related to a reduced sensitivity to their action [14–17].
Transcriptional factors
Recently three transcriptional factors, Oct-2, TTF-1, and EAP-1, have been identified as potential regulators of the cell network, which controls the GnRH secretion (“Upstream control”). They regulate the expression of genes involved in cell function and cell-cell communication (for details, see [18–21]. In the mammalian, hypothalamic lesions that induce sexual precocity activate both Oct-2 and TGF < 61537 > expression in astrocytes near the lesion site [18], suggesting that TGF < 61537 > is one of Oct-2 target.
The second candidate is TTF-1 (thyroid transcriptional factor-1), another homeobox gene. After birth, it remains expressed in selected neuronal and glial population of the hypothalamus. At the onset of puberty, TTF-1 enhances GnRH and erbB2 and KiSS-1 gene transcription but inhibits preproenkephalin promoter activity [19].
The third candidate is EAP-1, earlier known as C14ORF4. Like TTF-1, EAP-1 transactivates the promoter of genes involved in facilitating the advent of puberty while suppressing the expression of genes inhibitory to the pubertal process. Knocking down hypothalamic EAP-1 expression causes delayed puberty and disrupted oestrous cyclicity, both in rats and monkeys [20, 21].
KiSS–1/GPR54 system
Kisspeptin/metastin (KiSS-1) is a 53-amino-acid-peptide, earlier known as a suppressor of tumour metastases [22, 23]. The proteolitic cleavage of the primary KiSS-1 protein product originates the decapeptide kisspeptin-10 (KiSS-10), whose target is GPR54 receptor. GPR54-containing cells are diffusely distributed [24, 25]. Kiss neurons are important for the gonadal axis. They are located in discrete neuronal subsets of the preoptic area and the nucleus arcuatus [24, 26]. These cells include GnRH neurons and the anterior pituitary [27, 28].
The KiSS-1/GPR54 system has been recognized recently as the director of central functional network and peripheral signals. Genetic, physiological and clinical data strongly indicate that the KiSS-1/GPR system is an essential gatekeeper of GnRH function, and not just one more element in the cascade of signals controlling the gonadotropic axis. It allows the integration of central and peripheral inputs and plays therefore a decisive role in the control of reproductive function [29].
Both in rats and in primates, a marked increase in KiSS-1 and GPR54 mRNA levels coincide with the onset of puberty [24, 30]. Moreover, the sensitivity of GnRH system to kisspeptin is dramatically enhanced in adult versus juvenile mice [42]. Thus, the developmental activation of the GnRH axis by KiSS-1 at puberty reflects a dual phenomenon involving, not only the increase of kisspeptin tone, but also the enhancement of its efficiency to activate GnRH neurons, probably through post-transcriptional changes in GPR54 signalling [31].
Hypothalamic KiSS-1 system also plays an essential role in relaying the negative feedback input of sex steroids onto GnRH neurons. In male and female rats, bilateral gonadectomy evoked a consistent increase in KiSS-1 mRNA at the hypothalamus. Recent studies added further elements to the role of kisspeptin in the feedback control of gonadotropins by showing that negative regulation of hypothalamic KiSS-1 gene expression by oestrogen appears to be restricted to the nucleus arcuatus (Arc), known to be pivotal for negative feedback of sex steroids. In contrast, at the anteroventral periventricular nucleus (AVPN), KiSS-1 mRNA decreased after gonadectomy and increased after sex steroid replacement [32, 33]. AVPN is involved in mediating the positive feedback effects of oestrogen upon GnRH and LH surges. Therefore, via positive regulation of GnRH secretion, KiSS-1 neurons might be involved also in generation of the pre-ovulatory gonadotropin surge.
New strong evidence indicates that hypothalamic KiSS-1 may participate also in delivering information regarding the nutritional status of the organism to GnRH- neurons. Kiss-1 may therefore contribute to the link between energy stores and fertility [34, 35]. It has been shown that the permissive actions of leptin on the reproductive axis are mediated through modulation of GnRH secretion. Because GnRH neurons do not express leptin receptors, [35] kiss peptins might explain badly understood metabolic processes, signalled onto GnRH neurons via peripheral hormones such as leptin. However, several key aspects of the physiology of this system still remain open [36].
In conclusion, KiSS-1 system is an essential downstream element in the negative and (probably) positive feedback loops controlling gonadotropin secretion [37]. In addition, it may participate in the signalling to GnRH neurons of peripheral inputs from hormones such as leptin [38].
Leptin
Leptin is a 16-kDa peptide secreted by adipocytes. It is supposed to signal to the brain the critical amount of fat stores necessary for LHRH secretion, which in turn activates the hypothalamic-pituitary-gonadal axis [38]. Leptin was recently shown to suppress neuropeptide Y (NPY) expression in the nucleus arcuatus. NPY stimulates appetite, has an inhibitory effect on the gonadotropin axis and is involved with the inhibition of puberty in conditions of food restriction. Therefore, it has been hypothesized that leptin might exert its effects by acting on NPY. Under favourable nutritional conditions, the rise in leptin levels would suppress NPY, and in turn release the inhibitory effect of NPY neurons on the GnRH-LH/FSH axis, allowing the initiation of puberty [39].
On the other hand, there might be a direct peripheral negative effect of leptin on gonadal function through inhibition of the steroidogenic enzymes [39, 40].
In humans and animals, leptin blood concentrations rise with the onset of puberty. In adolescents of both sexes, the gradual rise in serum leptin levels before puberty together with a decline in circulating levels of soluble leptin receptor suggest that these changes may serve as one of the signals to the central nervous system that metabolic conditions are adequate to support pubertal development and trigger puberty [41].
No gender differences were detected in the relationship between leptin serum levels and fat mass in pre-pubertal and early pubertal subjects. In contrast, at Tanner stages IV and V, the serum hormone concentrations decrease in males and increase in females. In addition, a significant negative correlation between circulating concentrations of testosterone and leptin was described in males only [38].
Finally, it has been shown that normal leptin levels are necessary for the maintenance of menstrual cycles and normal reproductive function in adolescents of both sexes.
In conclusion, leptin seems to exert a positive central effect on the hypothalamic-pituitary-gonadal axis and a negative peripheral one on the gonads. Leptin might signal that the metabolic conditions are adequate to support pubertal development and trigger puberty.
Inhibin, activin, and follistatin
Inhibin and follistatin inhibit, and activin stimulates the expression, biosynthesis, and secretion of FSH [42–44]. They are synthesized mainly in the gonads. Inhibin, follistatin and activin are all three involved in the modulation of the hypophyseal-gonadal axis function. Inhibin and follistatin are both negative regulators of FSH secretion.
Inhibin, a heterodimeric glycoprotein, belongs to the TGF-b super family produced by ovarian granulosa cells. It is composed of an alpha and one or two beta subunits. These form two different products, inhibin A and B, respectively. FSH stimulates the synthesis and secretion of inhibins by the gonads, which in turn are involved in the feedback regulation of FSH secretion. In girls, inhibin A concentrations increase between stage 2 and 3 of puberty, remain constant throughout stages 4 and 5, and correlate positively with bone age, inhibin B and oestradiol serum levels [45, 46]. Inhibin B blood concentrations increase further similarly to inhibin A levels, reaching a plateau at 12–18 years. They correlate with oestradiol [45, 46] and FSH serum levels [48].
Blood concentrations of follistatin decrease slightly from stage 1–4 and 5 of puberty in girls [46]. Blood levels of activin A were shown to remain unmodified from stage 1–3 of puberty in females [48].
In conclusion, at puberty the concentrations of the two negative regulators of FSH secretion, inhibin and follistatin, change in opposite directions [46], whereas the blood levels of a positive regulator, activin A, increase, at least in females. All together, these alterations in serum concentrations of FSH-regulatory peptides lead to an increase in FSH secretion.
Melatonin
The marked increase in LH amplitude at night observed in early puberty occurs at the same time of melatonin secretion. On the other hand, precocious puberty associated with pineal tumours and due to ectopic secretion of gonadotropins is independent of melatonin [14]. The role of melatonin in puberty is questioned.
Other hormones
Growth hormone (GH), insulin, insulin-like growth factor (IGF)-I, and its major binding protein, IGFBP-3, normally rise at puberty [49]. The increase in growth hormone and IGF-I concentrations is probably responsible for most of the metabolic changes observed during puberty, including insulin-resistance, increased beta-cell response to glucose, and growth spurt. GH, and not androgens, may directly affect insulin sensitivity regulating the glucose-insulin homeostasis at the time of puberty [50]. Adiponectin, an adipocytokine with antidiabetic and antiatherogenic effects, were recently shown to progressively decline in parallel with pubertal development in boys [51]. It is inversely related to serum testosterone and dehydroepiandrosterone sulphate levels [51].
11.2.3 Pubertal Maturation of the Hypothalamo-Hypophyseal-Gonadal Axis (Fig. 11.5)
The hypothalamic-pituitary-gonadal axis undergoes an active phase during foetal and neonatal development and then enters a resting phase that lasts for the rest of childhood until puberty.
Puberty begins with an activation of the hypothalamic-pituitary-gonadal system. Changes in GnRH pulsatility during puberty are reflected by the peripheral LH- and FSH-Levels. Qualitative and quantitative changes in LH secretion resulting from pulsatile GnRH secretion, occur approximately 2 years before the appearance of secondary sexual characteristics. At puberty, LH pulsatile secretion is characterized by a 28-fold increase in the pulse amplitude, whereas pulse frequency increases only 1.8-fold. During prepubertal years, both LH and FSH secretions are preponderant during night-time. In the peripubertal period the secretion of gonadotropins increases during sleep, and stimulation with exogenous GnRH shows an enhanced release of LH from the pituitary gland that may be useful in differentiating a pubertal from a pre-pubertal response. Throughout puberty then, gonadotropin pulses further increase becoming apparent during daytime also.
Several studies have been published suggesting that the mechanisms underlying the onset of puberty are different in girls and boys, and different modes of transmission of induction of puberty in boys and girls were revealed [52]. Among other differences, in girls, FSH levels increase during the early stages, and LH levels during the later stages of puberty with a 100-fold increase in hormone concentrations. In contrast, in boys, FSH levels rise progressively through puberty with an increase in amplitude only, whereas LH levels increase in early puberty reaching a plateau shortly [14, 17].
11.2.4 Acceleration of Puberty
Puberty occurs today earlier than a century and even earlier than half a century or 20 years ago. In Tanner’s original report [1, 5] white girls had a mean age at onset of breast development and pubic hair of 11.2 and 11.7 years, respectively. The normal mean age at onset of pubertal characteristics in young girls has been revised in 1997 in a considerable population of 17,000 girls evaluated in a cross-sectional study [53]. It has been shown to vary with race, ethnicity, geographical location, and environmental and nutritional conditions.
Compared to Tanner’s original report, pubertal development appears to begin up to 1 year in advance in white and up to 2 years in African-American girls. Breast stage 2 is reported to occur in white girls at 9.96 ± 1.82 years (mean ± SD) with upper and lower limits of 7 and 13 years, and in African-American at 8.87 ± 1.93 years with limits between 6 and 13 years. Pubic hair would occur at 10.51 ± 1.67 and 8.78 ± 2.00 year in white and African-American girls, respectively [54]. In the US white girls puberty would begin by 10 years of age on average, and African American between 8 and 9 years [1, 5, 54, 55].
The age of menarche has been shown to decrease significantly since the nineteenth century. With respect to the first data published by Tanner [5] and Largo [4] 50 and 30 years ago, respectively, it continues to decrease. Menarche seems to occur earlier in white British girls (13.5 years) in 2004 [56] than in 1962 [5] and is reported to occur at 12.88 ± 1.2 years in white and at 12.16 ± 1.21 year in African-American girls [54]. In 2006, a large German survey found the median age at menarche to be 12.8 years [57], suggesting that the secular trend to an earlier menarche is continuing.
11.3 Delayed Puberty
11.3.1 Definition
Puberty is the period of life that leads to adulthood through complicated and sometimes painful physiological and psychological changes. Delayed puberty may have a dramatic impact on the mental and social development of an adolescent.
In the literature, different definitions for “delayed puberty” can be found.
The classical endocrinological definition and the current paediatric definition are identical for girls, but slightly different for boys:
Endocrinological definition (Grumbach and Styne [42])
Delayed puberty is defined as the absence of signs of puberty in healthy girls at age 13 years and in healthy boys at the age 13.5 years (2 SD above the mean age at start of puberty).
Paediatric definition: Delayed puberty is defined as the absence of signs of sexual maturation by an age more than 2–2.5 SD values above the mean of the population (traditionally breast development by 13 years in girls and testicular development by 14 years in boys) (Marshall and Tanner [1]; Lee [2]; Brämswig and Dübbers [55]).
11.3.2 Incidence
Delayed puberty is a rare condition, occurring in only approximately 2.5 % of the population. [1, 2, 42, 55]. The relative incidence of the different forms of hypogonadism in delayed puberty is shown on Table 11.1. In the series of Reindollar et al. [58], hypogonadotropic hypogonadism is found in 31 %, hypergonadotropic hypogonadism in 43 % and eugonadotropic hypogonadism leading to primary amenorrhea in presence of partial or complete development of secondary sex characteristics in 26 %.
Hypogonadotropic Hypogonadismus | 31 % |
Idiopathic | 10 % |
GnRH-deficiency | 7 % |
Anorexia | 3 % |
Other endocrinopathies | 4 % |
Organic | 13 % |
Hypergonadotropic Hypogonadismus | 43 % |
Abnormal karyotype | 26 % |
Normal karyotype | 17 % |
Eugonadotropic hypogonadism a | 26 % |
Rokitansky-Kuster and similar | 17 % |
Testicular feminization | 1 % |
11.3.3 When and How to Investigate?
There are no guidelines indicating when in the absence of pubertal signs an investigation should be started. Following both definitions listed above, in girls, a first evaluation should be done not later than at the age of 13.
Important is empathetic counselling to counteract the mostly deep anxiety due to the fact of being different from other girls at the same age. The child and the parents have to be fully and accurately informed and reassured that an underlying pathological process is rare and that the delay of the onset of puberty is mostly due to a benign, often familiar, deviation from the normal time course.
In most recommendations, a precise diagnostic evaluation is recommended in girls with persisting absence of the onset of puberty at the age of 14.5 years (mean + 3 standard deviations). However, a further evaluation is recommended earlier if a girl without onset of puberty starts to suffer because she becomes socially isolated among her classmates because of her physical retardation. Therefore, acceleration of puberty has to be taken into account for the decision when to start clinical evaluation in absence of pubertal signs.
In conclusion, investigation has to be started earlier than it has been recommended 20 years ago. It depends on the psychosocial pressure exerted on a child by the pubertal development of the pair group of schoolmates and friends.

Fig. 11.5
The determination of serum FSH, together with family history and clinical signs, allows in a simple way a first preliminary classification of girls sufferingfrom delayed puberty. Workup numbers relate to the chapters of this review
Figure 11.5 presents a simplified flow chart of the assessment of delayed female puberty. It describes schematically the process for the investigation of adolescent girls presenting with lack of spontaneous pubertal development. Shaded boxes show the major differential diagnoses of constitutional delay of growth and puberty, hypogonadotropic hypogonadism, and hypergonadotropic hypogonadism. However, no clinical algorithm can fully meet the requirements of all individual cases. Thus, adapted clinical decision–making is important at each stage.
11.3.3.1 Hormone Measurements
Gonadotropins
Basal levels of FSH and LH are low in patients with HH or constitutionally delayed puberty and elevated in hypergonadotropic hypogonadism.
Levels of FSH and LH remain low after one GnRH injection in hypothalamic and in hypophyseal hypogonadism.
Levels of LH and FHS increase in hypothalamic hypogonadism with intact pituitary function (but not in hypophyseal hypogonadism) after repeated pulsatile administration of GnRH (0.1 mg GnRH per injection).
When 0.1 mg of GnRH is injected, pubertal onset is characterized by LH/FSH >1.
Oestradiol
In girls, at pubertal onset, oestradiol levels are >40 ng/ml (<10 ng/ml before puberty).
Inhibin B and Anti-Müllerian Hormone (AMH)
The distinction between constitutional delay of growth and puberty (CDGP) and idiopathic hypothalamic or hypophyseal hypogonadism (IHH) is still a difficult clinical issue. Harrington and Palmer conclude that basal inhibin B may offer a simple, discriminatory test if results from recent studies are replicated: very low levels indicate a high likelihood of IHH [59]. However, current literature does not allow today for recommendation of any diagnostic test for routine clinical use. This applies, too, to the clinical use of AMH in the investigation of delayed puberty [60].
Other Hormones to be Checked
Pituitary deficits should be evaluated by measuring IGF-I, T4, TSH and cortisol.
11.3.3.2 Bone Age
A bone age <11 years in girls with growth failure is encountered in constitutionally delayed puberty.
Bone ages >11 years in girls require further investigation to eliminate hypogonadism.
11.3.3.3 Pelvic Abdominal Ultrasonography
In case of hypergonadotropic hypogonadism, gonads may be small or absent. At the onset of puberty, the ovaries develop follicular cysts long before menarche. Multicystic ovaries with more than six cysts are a normal phenomenon and are already observed in the early stages of puberty [61, 62]. At that stage, these normal cysts should not be confounded with an early expression of a later PSO-syndrome. If ovarian volume is >2 ml and the uterus >35 mm, puberty is imminent [63].
The uterine volume increases at first without, and then with, a visible layer of uterine mucosa. This mucosa layer is induced by the slowly increasing oestrogen secretion.
11.3.3.4 Karyotype
Independent of dysmorphic features suggestive of Turner syndrome, a karyotype should be performed in hypergonadotropic hypogonadism if the patient’s history (e.g., chemotherapy, X-ray treatment) cannot explain the gonadal pathology.
11.3.3.5 Brain Magnetic Resonance Imaging (MRI)
In presence of unexplained low levels of LH and FSH, organic pituitary or hypothalamic disease should be eliminated. MRI is the most efficient imaging examination. Agenesis of the olfactory bulbs is typical for Kallmann syndrome. Measurement of the pituitary and pituitary stalk is fundamental.
11.4 Impact on Fertility of the Different Forms of Delayed Puberty
11.4.1 Constitutional Delay of Growth and Puberty
Constitutional delay of growth and puberty (CDGP) is the most common cause of delayed puberty in girls with 30 % of cases, as it is in boys [66]. CDGP is defined as a delay of growth occurring in otherwise healthy adolescents with stature reduced for chronological age, but generally appropriate for bone age and stage of pubertal development, both of which are usually delayed. It is more frequent in boys than in girls with a 10:1 ratio and is the most common cause of delayed puberty (80–90 %).
In most cases delayed puberty is not due to any underlying pathology, but instead represents an extreme end of the normal spectrum of pubertal timing, a developmental pattern referred to as constitutional delay of growth and maturation [66]. The characteristically retarded linear growth occurs during the early years of life and is followed by regular growth paralleling the normal growth curve throughout the rest of prepubertal years. Pubertal growth spurt is attenuated and occurs after the usual expected time. In girls, exclusive maternal inheritance seems to be the major mode of inheritance whereas for boys the mode of inheritance is almost equally maternal, paternal or bilineal [52]. The majority of cases (70–80 %) are familial. Sedlmyer & Palmert classified family histories of pubertal timing among primary relatives in 95 of 122 of the CD and in 25 of 45 of the functional hypogonadotropic hypogonadism (FHH) cases. Analysis revealed at least a tendency to pubertal delay in 77 % of the CDGP and in 64 % of the FHH families and a diagnosis of delay in 38 % of the CDGP and 44 % of the FHH families. Both parents contributed to the positive family histories. The rates of positive family histories among the CDPD and FHH groups were approximately twice those seen among the other subjects in our case series [66]. Bone mineral density can be compromised by the low serum steroid concentrations measured [67, 68]. Specifically, the attainment of peak bone mass may be impaired, although recent data do not indicate significant changes in volumetric bone mineral density in young men with previous CDGP compared with appropriate controls [69].
The sleep-related increase in LH concentrations that characterizes the onset of puberty, is normally present in CDGP children. As a consequence of inadequate production of gonadal steroids, acute provocative tests may show a GH response wrongly consistent with partial GH deficiency [70]. Pre-treatment with oestrogens in girls results as expected in the normalization of the GH responses. The LH response to the LH-RH analogue leuprolide acetate is intermediate between that of hypogonadal patients and normal pubertal children, and is therefore useful in differentiating CDGP from hypogonadotropic hypogonadism. Recently, a critical appraisal of available diagnostic tests has been published [59].
Supportive care is essential. Although no specific treatment is required, the psychosocial problems faced by CDGP children may force physicians to substitute [55, 64, 71]. In girls, oestrogen therapy is recommended only after statural considerations have been carefully taken into account. Ethinylestradiol should be avoided. The administration of oestrogen, even in small amounts, leads to progressive skeletal maturation, and ultimately to epiphyseal fusion. The use of anabolic steroids or growth hormone to stimulate growth is highly controversial [72–75] and is not recommended in most reviews [66].
The inheritance patter of CDGP has been recently analysed by Winter et al. [52]. In girls, exclusive maternal inheritance seems to be the major mode of inheritance.
Impact on fertility
There are no published data suggesting that compared to children with normal puberty, fertility may be decreased in adulthood in individuals who had lived a constitutionally delayed puberty.
11.4.2 Other Forms of Hypogonadotropic Delay of Growth and Puberty
Table 11.1 lists the most important causes of delayed puberty other than constitutional delay of puberty and growth. These causes are usually grouped in four categories:
Delayed puberty due to congenital hypothalamic hypogonadotropic hypogonadism
Delayed puberty due to functional hypothalamic hypogonadotropic hypogonadism
Delayed puberty due to hypophyseal hypogonadotropic hypogonadism
Delayed puberty due to congenital or acquired hypergonadotropic hypogonadism
The characteristic endocrine pattern for hypothalamic hypogonadotropic, hypophyseal hypogonadotropic and hypergonadotropic hypogonadism is presented on Fig. 11.6.
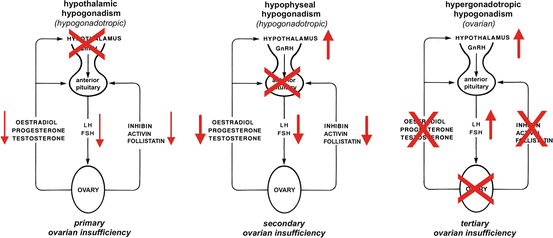
Fig. 11.6
Schematic presentation of the classical endocrine defects in primary, secondary and tertiary ovarian insufficiency
11.4.2.1 Hypothalamic Hypogonadotropic Hypogonadism (Primary Ovarian Insufficiency)
Delay of Puberty in Organic Hypothalamic Hypogonadotropic Hypogonadism (HH) (Table 11.2a)
Congenital (permanent) hypothalamic hypogonadism |
Isolated GnRH deficiency |
With ansomia (Kallmann syndrome) |
Without ansomia |
Associated with a syndrome such as |
Prader-Willi |
Laurence–Moon |
Bardet–Biedl |
etc. |
Acquired hypothalamic hypogonadism |
CNS tumours (cranyopharngeoma, germinoma etc.) |
Metastases from non-CNS-tumours |
Infections of the central nervous system |
Systemic infections, such as tuberculosis, syphilis, Trypanosomiasis |
Infiltrating processes ans storage diseases such as haemochromatosis (Thalassaemia major!), histocytosis, granulomas,sarcoidosis, M. Wilson |
Multiple sclerosis |
Head injury |
Stroke, rupture of anevrysm |
Cerebral surgery |
Chemotherapy/radiotherapy |
In girls, HH is proposed when plasma gonadotropins are normal or low, with lack of pubertal signs at 13 years of age. Puberty is absent or partial (congenital and early acquired forms), or arrested in an intermediate stage (acquired forms), depending on the appearance of the pathology in relation to the onset of puberty. Serum gonadotropin levels are low or inappropriately low-normal, sex steroids are low.
Isolated GnRH Deficiency
Congenital isolated hypothalamic hypogonadism (CHH) is clinically characterized by a partial or complete lack of puberty and a primary infertility due to a deficient GnRH-induced gonadotropin secretion, in the absence of anatomical abnormalities in the hypothalamic and pituitary region, and by normal basal and reserve testing of the remaining pituitary hormones.
Biologically, CHH is defined by low or normal serum levels of LH and FSH in the setting of low sex steroids. All other hypophyseal functions are normal as is the imaging of the hypothalamo–pituitary region. Patients with CHH typically present in adolescence or early adulthood with delayed onset of puberty, primary amenorrhea, poorly developed sexual characteristics, and/or infertility.
There exist two clinical variants of congenital GnRH deficiency: the form without anosmia and the GnRH deficiency with anosmia. When CHH is associated with anosmia or hyposmia it is termed Kallmann syndrome [76–78]. The Kallmann syndrome is the classic example of congenital hypothalamic hypogonadism. The first description of the so-called Kallmann syndrome has been published by de Morsier [79]. It is due to an impairment of the normal migration of the GnRH neurons from the region of the olfactory nerve to the ventral hypothalamus through the cribriform plate (Fig. 11.4). The clinical features of Kallmann syndrome are variable, with X-linked and autosomal-dominant and -recessive causes and variable penetrance described. Renal anomalies and syncynesia may exist. Its prevalence is 1/8000–1/10,000 in men and 1:50,000 in women. MRI confirms aplasia or hypoplasia of the olfactory bulbs.
Most cases of Kallmann syndrome seem to be sporadic as a consequence of mutations in at least two autosomal genes.
Mostly, genetic mutations are responsible for CHH [55, 64, 80, 81]. Mutations in the KAL1 gene on the short arm of the X chromosome (Xp22.3) are responsible for the X-chromosomal recessive form, while mutations in the FGFR1 (fibroblast growth factor receptor 1) gene on the short arm of chromosome 8 (8p11.2–p11.1) are responsible for the autosomal dominant form (e12). At present, in human, the only type of hypothalamic hypogonadism attributed to a single gene defect is the alteration of GPR54 [76] where KiSS-1 is involved (see above). Subjects with mutations in the human leptin receptor gene have no pubertal development. Table 11.2b lists the classical genetic mutations leading to permanent hypothalamic hypogonadism, with and without anosmia. Gene mutations with normosmic congenital hypogonadal hypodonadism are shown on Table 11.3. In all six listed mutations, heredity is recessive. This explains why expression of the anomaly is rare although the frequency of the abnormal gene GnRH1 in Europe is quite high (1/50) [80].
Migration disorder of the GnRH neurons (Kallmann syndrome) due to mutations in (e12): |
The KAL1 gene (chromosome Xp22.3) |
The fibroblast growth factor receptor 1 (FGFR-1) gene (chromosome 8p11.2–p11.1) |
The prokineticin 2 gene (e13) |
The prokineticin 2 receptor gene |
Nasal embryogenic LHRH factor (NELF) |
Disturbances of GnRH secretion without anosmia or hyposmia (e19) |
Mutations of the GnRH receptor gene |
Mutations of the leptin gene |
Mutations of the leptin receptor gene |
Mutations of the G-coupled protein receptor 54 gene (GPR54) (e18) |
Table 11.3
Genes responsible, frequency and phenotype in normosmic hypothalamic hypogonadism. Heredity is in all six listed mutations recessive [80]
Gene responsible of hypogonadism | Frequency of gene | Phenotype |
|---|---|---|
GnRH1 | Europe: 1/50 | Complete HH |
USA: 1/310 | ||
GnRH-R | 40 % of cases of familial normosmic HH | Complete HH |
Sporadic mutations: 6–17 % of cases of hypogonadism | ||
KiSS-1 | Rare, no sporadic mutations described | Severe gonadotropic deficiency, absence of puberty |
KiSS-R | Rare, sporadic or familial idiopathic HH: 26 cases from 9 different families described | Severe gonadotropic deficiency, absence of puberty |
TAC3 neurokinin B | Rare, no sporadic mutations described | 2 of the 4 sisters with TAC3 mutations had spontaneous pregancies, another has regular cycles and the forth had an early miscarriage |
TAC3-R | Rare, no sporadic mutations but rare variant described in sporadic cases | 6 of the 7 males and 4 of the 5 females demonstrated evidence for reversibility of their hypogonadism |
Additional developmental anomalies can occur with CHH including unilateral renal agenesis, synkinesia (mirror movements), cleft lip and/or palate, sensorineural hearing loss, dental agenesis, and skeletal malformations [81]. In some forms of CHH, additional defects are observed. These specific phenotypes are known as syndromes with CHH and additional abnormalities such as coloboma, heart defect, atresia of nasal choanae, retarded growth/development or genital abnormalities. The best known of these clinical syndromes are the Prader-Willi, the Laurence-Moon and the Bardet-Biedl syndrome. CHH and ear abnormalities up to deafness is known as the CHARGE syndrome [82, 83].
Delay of Puberty in Functional Hypogonadal Hypogonadism (Primary Ovarian Insufficiency)
It has been estimated that 10–20 % of all women suffer at least once in their life from functional hypothalamic disorders, mostly stress [17]. If such a functional disorder occurs before the normal age of puberty, puberty may be delayed.
Transient hypogonadal hypogonadism is seen in systemic conditions such as anorexia starting before or around puberty, excessive exercise (athletic triad) in pubertal girls, in severe chronic diseases of any origin, in malnutrition and in emotional deprivation [66, 84–88]. Sedlmeyer et al. [66] and other groups [84, 85] listed over 25 different underlying chronic diseases in their analysis of children investigated for functional delayed puberty. Among them, in addition to eating disorders and intense exercise, endocrine diseases (GH deficiency, hyperprolactinemia (see above), hypothyroidism), diabetes mellitus, cystic fibrosis, Crohn’s disease, celiac disease, severe asthma, nephrotic syndrome, rheumatoid arthritis, systemic lupus erythematodes, sickle cell disease and thalassemia major, congenital heart disease, focal segmental glomerulosclerosis, glycogen storage disease type 1A, several oncological diseases (Hodgkin’s disease, leukaemia etc.), CNS disorders (particularly seizure disorders) and poor nutrition.
Acquisition of fat mass is involved in pubertal development. During starvation, in stress-induced amenorrhea with weight loss, in subjects with anorexia nervosa, and in strenuously exercising athletes, leptin and E2 levels fall concomitantly. By limiting the apposition of adipose tissue, chronic diseases affect the development of puberty and fertility by the same mechanism relayed through the hypothalamus, apart from the specific impact of their molecular alteration. As the effect of the drugs used to treat chronic diseases (e.g., corticosteroids) are undistinguishable from the chronic disease itself, pharmacological side effects have to be considered, too [85].
As long as these conditions persist, the onset of puberty remains blocked or its normal continuation stays arrested. In severe cases, a functional regression to prepuberty equivalent with the prepubertal quiescence of the ovarian axis may occur (see Fig. 11.4).
11.4.2.2 Delay of Puberty in Hypophyseal Hypogonadotrophic Hypogonadism (Secondary Ovarian Insufficiency)
Congenital or permanent hypophyseal hypogonadism is rare (Table 11.4). Intracranial tumour is a common cause of acquired hypogonadism in adolescence. Among these, craniopharyngeoma, a typical CNS tumour in adolescents, may lead to destructions in the hypothalamo-hypophyseal region [89]. If the pituitary stalk is compressed which is not rare in extrapituitary tumours such as craniopharyngeomas or metastases from non-CNS-tumours, other hypothalamo-hypophyseal axes in addition to the gonadal axis are affected. Neurosurgery for craniopharyngeoma is mostly followed by radiotherapy. In some other tumours, too, surgical resection may be complemented with radiotherapy and/or chemotherapy leading to secondary damage [90, 91].
Table 11.4
Classical causes of hypophyseal hypogonadotrophic hypogonadism (secondary ovarian insufficiency)
A. Congentital or permanent hypophyseal hypogonadism |
Classical congenital forms are: |
Isolated LH and FSH deficiency (“idiopathic isolated gonadotropin deficiency”) |
Panhypopuitarism (complete or partial) |
Congenital (genetic, “idiopathic”) |
Associated with a lesion of the midline/Rathke’s pouch |
Syndromes, such as CHARGE syndrome: combined pituitary hormone deficiency (coloboma, heart defect, atresia of nasal choanae, retarded growth/development, genital abnormalities, and ear abnormalities/deafness) |
B. Acquired hypophyseal hypoginadotropic hypogonadism |
Panhypopituitarism (partial or complete) |
CNS tumours, such as craniopharyngioma, hamartoma, germinoma etc. |
Metastases from non-CNS-tumours |
Prolactinomas |
Non-prolactin secreting pituitary adenomas |
Hypophysitis |
Infections, such as tuberculosis, syphilis, trypanosomiasis |
Sarcoidosis |
Eosinophilic granuloma |
Haemochromatosis (Thalassaemia major!) |
Multiple sclerosis |
Trauma |
Chemotherapy/radiation therapy |
In presence of an adenoma of the pituitary including makroprolactinoma, hypogonadotropic hypogonadism can result from the compression of pituitary tissue. In the case of prolactinoma or Cushing’s disease, delayed puberty may be secondary to the inhibition of GnRH secretion by the hormones secreted by the endocrine active hypophyseal adenoma, even it is small.
A rare cause of hypophyseal hypogonadotropic hypogonadism is the empty sella syndrome. Primary ES occurs when CSF enters the sella through a rent in the sellar diaphragm that may or may not be associated with increased intracranial pressure. Secondary ES is a result of an injury to the pituitary itself or the consequence of surgical or radiation treatment. The incidence of ES in children varies greatly depending on the population surveyed, ranging from 1.2 % (children without endocrine symptoms) to 68 % (children with known endocrinopathy) in the survey of Lenz and Root [92].
In adenomas of the pituitary, in empty sella and in craniopharyngeoma, clinically, visual disturbance or headaches may accompany pubertal arrest. It is essential that all patients with intra- or extrahypophyseal tumours undergo a complete evaluation of anterior and posterior pituitary function.
11.4.2.3 Impact on Fertility
Hypogonadotropic hypogonadism due to congenital hypothalamic disorders have very rarely and only in very light partial forms the chance to get later spontaneously pregnant. However, with the adequate treatment, the possibility to live later a normal pregnancy is excellent even in complete forms of hypothalamic hypogonadism (see below).
Hypogonadotropic hypogonadism resulting from hyperprolactinaemia can be treated medically by dopamin agonists [93]. Because the normalization of prolactin secretion by dopamin agonists allows not only the onset of normal pubertal development but also the uptake of a normal fertility, adolescents have to be informed that in case of intercourse without the desire of a child they need an adequate and efficient contraception.
In non–prolactin–secreting adenomas of the pituitary and in most other CNS tumours, surgical intervention is the usual first line treatment [94–96], followed frequently by radiotherapy or chemotherapy. These treatments per se may lead in survivors to permanent hypogonadism [90, 91]. Later spontaneous fertility depends on the destructions left by the tumour itself or by its treatments. As long as the ovaries are intact and have not suffered by chemotherapy or radiotherapy, the chances to become pregnant through ovulation induction remain intact.
In women, where the delay of puberty has been due to functional hypothalamic hypogonadism, the successful treatment of the underlying disease decides on later fertility. Particularly, in women with eating disorders, a complete remission is the conditio sine qua non if normalization of fertility is intended. However, the few longitudinal studies on later fertility show that the risk of a subnormal fertility pattern remains increased, as it has been observed in the “Avon Longitudinal Study of Parents and Children Fertility and prenatal attitudes towards pregnancy in women with eating disorders” [97]. In this study, Singleton and live births were included across four groups of women suffering from lifetime eating disorders:
Lifetime anorexia nervosa (AN; n = 171)
Lifetime bulimia nervosa (BN; n = 199)
Lifetime anorexia nervosa and bulimia nervosa (AN + BN; n = 82)
General population (n = 10,636).
The results show that women with AN (OR 1.6, 95 % CI 1.1–2.5; P < 0.021) and women with AN + BN (OR 1.9, 95 % CI 1.1–3.4; P < 0.020) were more likely to have seen a doctor for lifetime fertility problems than women from the general population. Furthermore, women with AN + BN were also more likely to take > 6 months to conceive (OR 1.9, 95 % CI 1.0–3.5; P < 0.04) and to have conceived the current pregnancy with fertility treatment.
All eating disorders groups experienced more frequently negative feelings upon discovering their pregnancy. Negative feelings remained still higher in the AN + BN group at 18 weeks of gestation. Finally, in spite of the longer time the AB women needed to get pregnant, unplanned pregnancies were more common in the AN group compared with the general population. This points to the persistence of an increased ambivalence against pregnancy in women with eating disorders. These last two findings have been confirmed by a second study [98, 99].
Women with lifetime AN had a higher prevalence of twin births compared with those without the disorder (3.5 versus 1 %), as did women with BN and women with AN + BN, albeit to a lesser extent [99]. All eating disorders taken together were associated with increased odds of having twins (OR 2.7, 95 % CI 1.0–7.9; P = 0.06). These associations persisted after adjustment for potential confounding factors such as lifetime AN, OR 2.7, 95 % 1.0–8.0, lifetime BN, OR 2.7 (95 % CI 1.1–6.4) and lifetime AN + BN, OR 3.9, (95 % CI 1.3–11.1). Interestingly enough, women with other lifetime psychiatric disorders had similar odds as women without psychiatric disorders.
11.4.2.4 Profertile Measures: Ovulation Induction in Hypogonadotropic Hypogonadism (Primary and Secondary Ovarian Insufficiency)
In the absence of the uptake of normal menstrual cycles, as it is the case in all forms of delayed puberty with permanent hypogonadal hypogonadism, ovulation induction should be used to induce pregnancy. It has to be stressed that the administered hormones have to be considered and handled as a substitution. Therefore, the lowest efficient dose of GnRH/pulse or of HMG resp. FSH/LH per day has to be used to obtain a monofollicular response of the ovary.
Related posts:
 Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
 Management of Peripheral Precocious Puberty in Girls
Management of Peripheral Precocious Puberty in Girls
 Adolescent Pregnancy and Contraception
Adolescent Pregnancy and Contraception
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


