First-line
Second-line (if first-line abnormal)
Clinical assessment
Height
Weight
Sitting height
Body mass index (BMI)
Tanner pubertal stage
Bone age
Endocrine biochemistry
IGF-1/IGF-BP3
GH provocation test (e.g. insulin tolerance test, glucagon stimulation test)
LH, FSH, oestradiol/testosterone (if <2 years, and >8 years in girls, >9 years in boys)
GnRH stimulation test
TSH, free T4 ± free T3
7, 8, 9 a.m. cortisol and ACTH (before dexamethasone)
Adrenal stimulation test (e.g. insulin tolerance test, standard or physiological synacthen test)
Early morning paired urine and plasma osmolalities and electrolytes
Water deprivation test
Tumour markers
PRL
AFP
β-hCG
Endocrine tests prior to any definitive treatment are crucial for several reasons. Firstly, differentiating craniopharyngiomas from other suprasellar tumours in this region such as germinomas (some of which secrete α-fetoprotein (AFP) and/or β-human chorionic gonadotrophin (β-hCG) ), prolactinomas (PRL) and somatotrophinomas (GH) is important as tumours such as germinomas and prolactinomas can be treated medically by chemotherapy and dopamine agonists, respectively, without necessitating biopsy or surgical intervention. Measurement of these plasma hormones (AFP, β-hCG, PRL, and GH or IGF-1), and occasionally cerebrospinal fluid AFP and β-hCG concentrations is therefore mandatory in all such cases.
A basal pituitary hormone screen helps identify which patients need prioritising for dynamic endocrine testing prior to any definitive surgery. Further dynamic testing should prioritise establishing the status of the hypothalamo-pituitary-adrenal and ADH axes prior to embarking on any surgical intervention in order to avoid the fatal consequences of uncorrected cortisol insufficiency and/or central diabetes insipidus . Pre-operative early morning paired urine and plasma electrolytes and osmolalities, as well as early morning cortisol and ACTH concentrations, should be measured particularly prior to the administration of high-dose dexamethasone for peritumoural oedema as this will subsequently suppress the hypothalamo-pituitary-adrenal axis. Morning cortisol values <200 nmol/l may indicate ACTH deficiency and in the absence of dexamethasone prophylaxis should be confirmed by insulin-induced hypoglycaemia (which will concurrently assess GH reserve) or standard synacthen stimulation, with a peak cortisol concentration of >500 nmol/l being normal.
Further investigation for other hypothalamo-pituitary endocrine deficits and their treatment must be supervised by a paediatric endocrinologist. Dynamic testing and treatment of GH and gonadotrophin deficiency can usually be delayed to the post-treatment period. It would, however, seem sensible to render a hypothyroid patient euthyroid pre-operatively. The pattern of endocrine dysfunction at diagnosis may also be important—for instance, isolated central DI is unusual as a presenting feature in craniopharyngiomas (usually only being present with other pituitary hormone deficiencies), and is usually a feature of germinomas and Langerhans cell histiocytosis [24, 25].
Endocrine Considerations in Perioperative Management
In the absence of dexamethasone prophylaxis and where a patient’s adrenal status has not been established, intramuscular or intravenous hydrocortisone in stress doses (1–2 mg/kg) should be administered at induction of anaesthetic, with repeated doses every 6–8 h thereafter. This should be gradually weaned to oral maintenance doses of hydrocortisone (10–15 mg/m2/day) over 5–10 days until adrenal status can be assessed.
Pre-operative central DI warrants desmopressin administration prior to surgery (0.1–0.2 μg intramuscularly/subcutaneously) with careful attention to fluid balance monitoring with an indwelling urinary catheter intra-operatively. Fluid losses should be replaced ml for ml with an additional 300 ml/m2/day to account for insensible losses. Uncontrolled posterior pituitary dysfunction is a significant risk factor for mortality and was found to be associated with nearly 50% of deaths from hypothalamo-chiasmatic low-grade gliomas, another suprasellar tumour [26]. Close monitoring by an experienced paediatric endocrinologist is essential as dangerous post-operative swings in salt and water balance resulting in rapid shifts from hyper- to hyponatraemia can occur for several reasons leading to the attendant risks of seizures and cerebral oedema.
Firstly, a well-described triphasic response of transient DI (up to 48 h), SIADH (up to 2 weeks) and a second phase of DI may evolve over the first 2–3 post-operative weeks. The latter is more likely to be permanent if it persists beyond 21 days, is preceded by a prolonged or severe period of SIADH, or if extensive surgery has been performed [27, 28]. This triphasic phenomenon is more frequent in children than adults (23% vs. 14% in craniopharyngiomas) [29]. The three phases may also occur independently, with or without the coexistence of cerebral salt-wasting syndrome which further complicates diagnosis and management. Additionally, changes in dosing of glucocorticoid replacement therapy, particularly when weaning from supraphysiological doses, may result in changes in requirement for desmopressin replacement. Other factors that increase the risk of posterior pituitary dysfunction include the use of anticonvulsants such as carbamazepine and lamotrigine, which can potentiate the effect of desmopressin and cause an SIADH-like syndrome. Lastly, surgically-induced damage to the thirst centre in the hypothalamus can lead to hypothalamic adipsia , where the child may need to be restricted to a set amount of fluid per day to maintain normal fluid balance.
Long-Term Endocrine Dysfunction in Craniopharyngioma Survivors
The pattern of post-treatment endocrine dysfunction seen in survivors of craniopharyngioma is remarkably very similar in frequency and timing, regardless of the treatment modalities used. Deficiency of GH is commonest (20–99%), followed by TSH/TRH (39–97%), LH/FSH (30–95%), ACTH (39–89%) and finally ADH (42–94%), and 31–84% of patients have panhypopituitarism [2–4, 6, 7, 30–38]. Longitudinal comparisons indicate that the hierarchical evolution of deficits mimics that of many other suprasellar tumours, with the GH axis being the most susceptible, and the ACTH and posterior pituitary axes being the most robust [3, 5, 26, 33, 39]. The frequency of deficits also mimics that of congenital neurodevelopmental malformations in this region such as septo-optic dysplasia [40]. Although it is well known that the various hypothalamo-pituitary axes are differentially sensitive to irradiation [41], the similar pattern seen even in its absence as well as with other suprasellar tumours and congenital disorders suggests that this observation is more to do with tumour position and the inherent susceptibility of various hypothalamo-pituitary networks to damage rather than the therapeutic modality per se. The continued possibility of new endocrine deficits emerging over time (Fig. 1) means that lifelong endocrine follow-up with regular clinical and biochemical assessments is crucial, with at least 6-monthly follow-up appointments until adult height is achieved, after which annual reviews should suffice with clear plans for transition to adult care.
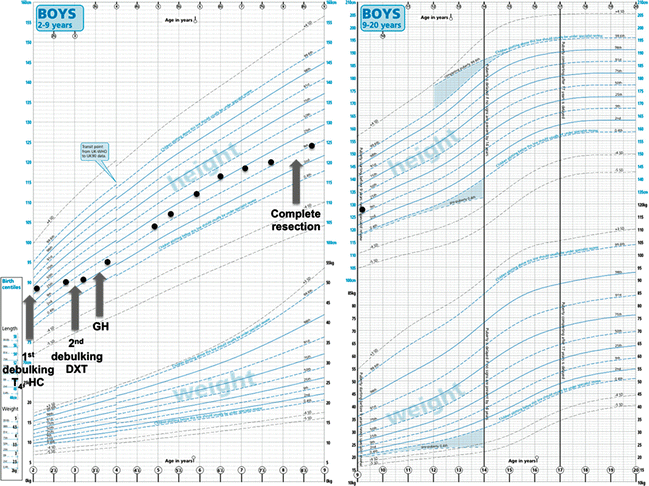
Fig. 1
Typical growth chart of a patient with craniopharyngioma who had multiple progressions and required multiple surgical interventions and radiotherapy. Not noted here for clarity are the multiple cystic progressions which required intracystic drain and reservoir insertions. Note the deceleration in height velocity prior to each progression. T 4 , levothyroxine; HC, hydrocortisone; GH, growth hormone
GH Deficiency
GH deficiency occurs in nearly all craniopharyngioma survivors at some stage. If not already present at diagnosis, it is virtually guaranteed to occur after any therapy to the hypothalamo-pituitary region. It has been shown that the GH axis is particularly sensitive to irradiation [26, 41, 42], and neurosecretory dysfunction with disturbances to spontaneous GH pulsatility may be present before eventual evolution to frank GH deficiency [43]. Diagnosis requires dynamic endocrine testing with the gold standard being the insulin tolerance test, but this is contraindicated in patients with a history of seizures resulting from tumour- or treatment-related damage due to the unavoidable side effect of hypoglycaemia. If performed, we would recommend routine glucose rescue at 25–30 min supported by an age-appropriate dose of emergency hydrocortisone at the end of the test, regardless of whether hypoglycaemia is achieved or not, due to the risk of concurrent ACTH deficiency. Other alternatives include the glucagon stimulation test , clonidine stimulation test and arginine-GHRH stimulation test, although the latter will not detect GH deficiency resulting from hypothalamic damage and disruption of GHRH release. As mentioned previously, serum IGF-1 and IGF-BP3 are less accurate markers of GH deficiency, with the former being also dependent on nutritional status and liver function, but may be useful in cases where dynamic testing is considered too hazardous.
Paradoxically normal growth may also occur despite GH deficiency, either due to the phenomenon of “growth without growth hormone ”, which is poorly understood and thought to be secondary to hyperinsulinaemia occurring from rapid post-treatment weight gain [44], or precocious puberty. It is worth noting that GH therapy in replacement doses is not associated with an increased risk of tumour recurrence or progression in both craniopharyngiomas [2, 45, 46] and other suprasellar lesions [26]. There may, however, be an increased risk of second primary neoplasms but the evidence for this is not completely clear [47]. Given this, we tend to advocate “minimum effective doses” for GH replacement, starting at a dose of 0.5–0.7 mg/m2/day (10–15 IU/m2/week) and aiming for normal IGF-1 concentrations and an age-appropriate height velocity.
Gonadotrophin Dysfunction
Gonadotrophin dysfunction can manifest either as central precocious puberty (defined as a testicular volume of ≥4 ml in a boy <9 years or breast budding in a girl <8 years), pubertal delay (failure to enter puberty by age 14 in a boy and 13 in a girl), or arrest (manifesting as sexual dysfunction or infertility in boys and primary amenorrhoea or oligomenorrhoea in girls). Central precocious puberty tends to occur in the setting of irradiation exposure and is increased in younger children and with lower irradiation doses (thought to be secondary to disruption of inhibitory hypothalamic pathways on the GnRH pulse generator) [43, 48]. As previously mentioned, the coexistence of precocious puberty with GH deficiency may mask growth failure, and height velocity must always be assessed in light of a child’s pubertal stage and bone age. Treatment with GnRH analogues such as triptorelin alongside GH supplementation in the presence of deficiency can help restore adult height if commenced early, but consideration must be given as to whether delaying puberty is appropriate for each individual child.
Conversely, pubertal delay or arrest as a result of hypogonadotrophic hypogonadism can result from either tumour- or treatment-related damage to the hypothalamus or pituitary. The possibility of pubertal arrest means that even patients with a spontaneous pubertal onset must be followed up carefully. The concurrent presence of untreated GH deficiency (which is usually inevitable in this scenario) can be managed by commencing GH supplementation at least 6 months prior to sex steroid supplementation. The timing of sex steroid initiation, however, must be tailored appropriately to the individual child, bearing in mind that delaying commencement beyond the usual pubertal age (~12 years in girls and 14–15 years in boys) is probably inappropriate, particularly given the long-term benefits of sex steroid replacement on bone mineral accretion and long-term bone health [49–51].
In both cases, a GnRH stimulation test provides supporting information but is not always necessary. It is also worth noting that central precocious puberty and later hypogonadotrophic hypogonadism are not mutually exclusive entities. Indeed, in a retrospective longitudinal analysis of survivors of childhood hypothalamo-chiasmatic low-grade gliomas , Gan et al. [26] demonstrated that children with central precocious puberty were potentially at increased risk for future hypogonadotrophic hypogonadism. Consequently, children who have been treated for the former with GnRH analogues should be monitored carefully on the cessation of such treatment, looking out for failure of subsequent normal pubertal progress.
TSH/TRH Deficiency
Unlike the thyroid gland itself, TRH-secreting neurons and thyrotrophs in the pituitary gland are relatively resistant to irradiation [41]. Secondary and/or tertiary hypothyroidism is therefore only likely to arise in the setting of hypothalamic involvement of the tumour or from direct surgical damage. As previously mentioned, TRH stimulation tests do not aid in the differentiation between secondary and tertiary hypothyroidism and serial thyroid function tests are more specific and easier to perform [21]. In this setting, dose titration of levothyroxine supplementation should be based entirely on free thyroxine concentrations and not TSH. Some clinicians favour keeping the free thyroxine concentration in the upper half of the normal range, given the predisposition of many craniopharyngioma survivors to obesity.
ACTH Deficiency
Out of all the anterior pituitary hormones, the hypothalamo-pituitary-adrenal axis is fortunately most robust to irradiation. Post-operative assessment of this axis after the use of perioperative dexamethasone and subsequent maintenance hydrocortisone replacement is probably best left for at least 3 months. If neither dexamethasone nor hydrocortisone has been commenced, then serial monitoring of early morning cortisol concentrations (aiming for >200 nmol/l) should be performed.
The gold standard insulin tolerance test assesses the entire hypothalamo-pituitary-adrenal pathway and concurrently tests the integrity of the GH axis as well, but may not always be possible as discussed above. Given the risks associated with hypoglycaemia resulting from this test, it should be performed in a dedicated paediatric endocrinology centre used to managing patients with multiple pituitary hormone deficiencies. Other methods of assessment such as the standard (high-dose) synacthen test, low-dose synacthen test and 24-h cortisol day curves are less reliable but may be useful in cases where the insulin tolerance test is contraindicated. It is worth remembering that the standard synacthen test involves a supraphysiological stimulus of synthetic ACTH and may be less sensitive at detecting more subtle degrees of deficiency [52–54]. However, the sensitivity of the low-dose synacthen test is also widely debated, not least because the protocols used in different studies vary considerably [55–57]. Regardless of the method used, hydrocortisone should be omitted on the morning of the test as it interferes with most commonly used biochemical assays for cortisol.
Hydrocortisone replacement therapy is generally given at a dose of 10–15 mg/m2/day, with parents and/or patients advised to double or even treble their doses in the face of illness. Doses can be titrated to trough concentrations, or more accurately using a 24-h cortisol day curve. More recently, we have also advised patients to take an extra double dose of hydrocortisone at 4 a.m. when they are unwell, as this coincides with the beginning of the physiological cortisol morning surge. Patients should also be educated on how to administer emergency intramuscular hydrocortisone (doses <1 year 25 mg, 1–5 years 25–50 mg, >5 years 100 mg) and correct hypoglycaemia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


