Fig. 8.1
Proposed model of taxane mechanism of action in prostate cancer. AR associates with microtubules and translocates to the nucleus via the motor protein dynein. Taxanes inhibit depolymerization of microtubules and block microtubules dynamics. By interfering with microtubule dynamics, taxanes cause a cell cycle arrest in the G2/M phase, and inhibit AR nuclear translocation as an additional mechanism of action in mCRPC. The mechanisms of action of enzalutamide (MDV3100) and abiraterone are also shown. Enzalutamide exerts its effect by inhibiting AR nuclear translocation, DNA-binding and co-activator recruitment. Abiraterone inhibits androgen biosynthesis by irreversibly blocking CYP17A1, a crucial enzyme in steroidogenesis. Reprinted by permission from the American Association for Cancer Research: Thadani-Mulero M. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Research. 2012;72(18):4611–5
Following phase I/II studies yielding PSA responses, pain responses, and objective tumor responses for docetaxel [9, 10], two large phase III trials TAX327 and SWOG 99-16 were initiated [11, 12]. TAX327 was conducted in 1,006 men with mCRPC who were randomized to receive 3-weekly docetaxel (75 mg/m2), weekly docetaxel (30 mg/m2), or 3-weekly mitoxantrone (12 mg/m2), each with prednisone [11]. OS of patients who were treated with docetaxel in the 3-weekly regime was superior as compared to mitoxantrone with an OS of 19.2 vs. 16.3 months (HR 0.79, 95 % CI 0.67–0.93) in the final analysis [13]. The docetaxel 3-weekly arm also showed better palliation, with more patients having pain (35 vs. 22 %, p = 0.01) and quality of life responses (22 vs. 13 %, p = 0.009) as compared to mitoxantrone. The docetaxel weekly schedule showed a trend towards improved OS, but did not reach statistical significance. The TAX327 updated survival analysis also contained a post-hoc analysis which demonstrated that the trends in OS were consistent among several subgroups of patients based on age (<68 vs. ≥68 years), pain vs. no pain at baseline, and baseline PSA <115 vs. ≥115 ng/mL.
Neutropenia was the most common observed grade 3/4 toxicity and occurred more frequently in patients receiving 3-weekly docetaxel (32 %). Despite the high incidence of neutropenia, febrile neutropenia was rare (3 %) and other grade 3/4 toxicities all occurred in less than 5 %.
A second trial, SWOG 99-16 was designed on the assumption that the combination of docetaxel and estramustine had the greatest therapeutic potential. Seven-hundred and seventy patients were randomized to receive 280 mg estramustine three times daily on days 1–5, plus docetaxel 60 mg/m2 on day 2, preceded by 60 mg of dexamethasone divided in three doses, or mitoxantrone 12 mg/m2 on day 1 plus 5 mg of prednisone twice daily [12]. Both were given in a 21-day cycle, and dose escalation to docetaxel 70 mg/m2 or mitoxantrone 14 mg/m2 was allowed in cycle 2 if no grade 3/4 toxicities were observed during the first cycle. Median OS was superior in the group receiving docetaxel as compared to mitoxantrone (17.5 vs. 15.6 months, respectively), with an HR of 0.80 (95 % CI 0.67–0.97). The group treated with docetaxel and estramustine had significantly higher rates of grade 3 and 4 neutropenic fever (5 vs. 2 %), cardiovascular events (15 vs. 7 %), and nausea and vomiting (20 vs. 5 %), as compared with the group treated with mitoxantrone and prednisone.
Taken together, the results of these two phase III studies showed that docetaxel in a 3-weekly regimen improved OS, which was the primary endpoint of both trials. Weekly docetaxel did not appear to be better tolerated than the 3-weekly regimen, and showed only a trend towards better efficacy as compared to mitoxantrone. The SWOG study did not reveal greater benefit by the addition of estramustine. Because of the lack of superior activity and greater toxicity by the addition of estramustine, docetaxel every 3 weeks plus low-dose prednisone subsequently became the standard of care for patients with mCRPC [14].
In multivariate analysis of TAX327, a total of ten independent prognostic factors for survival were identified including the presence of liver metastases, number of metastatic sites, clinically significant pain, Karnofsky performance status, type of progression, pretreatment PSA doubling time, baseline PSA, tumor grade, baseline alkaline phosphatase, and baseline hemoglobin [15]. These prognostic factors have been elaborated into a nomogram (Fig. 8.2). Such decision making tools are informative, can facilitate tailoring of therapy, and can simplify important clinical decisions such as when to start cytotoxic chemotherapy. Although the survival benefit obtained by docetaxel compared with mitoxantrone is consistent among patients with and without pain at baseline (HR 0.73 and 0.85, respectively), there is a substantial difference in OS time (14.4 months for patients with pain vs. 21.3 months for patients without pain). However, this does not necessarily imply benefit from the early use of chemotherapy, but may rather guide treatment in asymptomatic patients by defining patients at greater risk of imminent disease progression and death. These patients may be candidates for chemotherapy, even in the absence of symptoms.
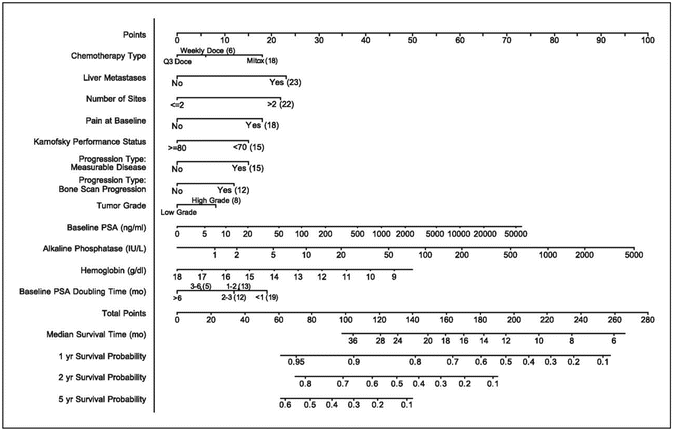
Fig. 8.2
Nomogram for survival of patients with progressive mCRPC, including data derived from 686 patients and 518 mortality events. Note: A present pain intensity of ≥2 and/or an analgesic score of ≥10 were defined in the original protocol as indicative of the presence of significant pain. Instructions for physician: Locate the liver metastasis axis. Draw a straight line upward to the points axis to determine how many points toward survival the patient receives for the presence or absence of liver metastases. Repeat this process for each predictor variable and sum the points for each predictor. Locate this sum on the total points axis. Draw a straight line downward from the total points axis to identify the predicted median survival and the predicted 1-, 2-, and 5-years predicted overall survival probabilities. Instructions to patient: “Mr. X, if we had 100 men exactly like you, we would expect < nomogram prediction × 100 > to be alive in 1, 2, and 5 years, respectively, and we expect 50 of those to be alive after < median survival prediction > months.” Reprinted by permission from the American Association for Cancer Research: Armstrong A. A contemporary prognostic nomogram for men with hormone-refractory metastatic prostate cancer: a TAX327 study analysis. Clinical Cancer Research. 2007;13(21):6396–403
Nonetheless, in the TAX327 study a decrease in quality of life was more often observed in patients with minimal symptoms at the start of chemotherapy [16]. Therefore delaying chemotherapy may be a suitable approach in patients with minimal symptoms. Those patients who have no symptoms yet, but are more likely to develop symptoms in the near future due to bone scan progression and/or the development of anemia should be considered candidates for docetaxel chemotherapy [14].
With the recent FDA and EMA approval of the CYP17 inhibiting agent abiraterone in the pre-docetaxel setting, it has become increasingly important to identify subgroups of patients who may have greater benefit by the use of chemotherapy in order to better tailor treatment choices. Recently, Azria et al. reported a high Gleason score [8–10] at the time of diagnosis to be an independent risk factor for poor response to abiraterone [17, 18]. In addition, a retrospective analysis of patients with mCRPC enrolled in clinical trials demonstrated that patients who had a short response to prior androgen deprivation therapy (ADT) (<16 months) had poor PSA responses and PFS when treated with secondary hormonal therapies such as abiraterone and enzalutamide [19]. In this light, a recent post-hoc analysis of the TAX327 study was conducted which revealed that the survival benefit obtained with docetaxel as compared to mitoxantrone was most pronounced in patients with high Gleason score tumors (Gleason 7–10) [20]. Furthermore, two prospective databases of patients with mCRPC demonstrated similar PSA responses and clinical benefit obtained by docetaxel, irrespective of the duration of response to ADT [21].
In an era of shifting paradigms in mCRPC with abiraterone becoming available also prior to docetaxel chemotherapy, Gleason score and prior response to ADT may serve to discriminate between patients who benefit most from docetaxel chemotherapy as first-line treatment. Docetaxel seems to exert efficacy particularly in high Gleason score tumors irrespective of response to ADT. In contrast, in patients with better differentiated tumors and durable responses to ADT abiraterone might be a good treatment option. In the future, these observations should be prospectively validated in order to further personalize first-line treatment options for patients with mCRPC.
Mechanism of Action of Taxanes: Emerging Data on AR Transport as Part of Their Efficacy
As mentioned above, docetaxel and cabazitaxel also impair AR signaling, which in the setting of mCRPC might in fact be responsible for part of the therapeutic efficacy [6–8]. Recently, clinical and preclinical evidence is emerging about potential cross-resistance between docetaxel and abiraterone [8]. A clinical report on patients treated with docetaxel who had previously been treated with abiraterone showed an OS of only 12.5 months, which was significantly less than the 19 months predicted for this patient population [22]. Moreover, PSA declines ≥50 % were observed in 26 % of patients, compared to 45 % in the TAX327 study [11], and no responses to docetaxel were observed in abiraterone-refractory patients. A likely explanation is that antitumor activity of taxanes in mCRPC is partly depending on its impact on AR signaling. When patients are treated with abiraterone first, it could very well result in impaired effectiveness of docetaxel due to annulling its effects on the AR. Hence the sequence of abiraterone followed by docetaxel upon progression could result in decreased effectiveness of the chemotherapy and thus impair the eventual clinical benefit.
Cabazitaxel, however, seems to retain activity in the third-line setting following docetaxel and abiraterone, with ≥50 % PSA declines in 42–49 % of patients [23, 24]. Prospective clinical studies should further define the implications for the optimal treatment sequence of these treatment options for patients with mCRPC.
Docetaxel Retreatment
Sooner or later all patients will progress during or after treatment with docetaxel. Patients who relapse after an initial response to docetaxel may again respond to a second or even third series of docetaxel cycles [25–27]. Since the phase II data on docetaxel rechallenge have been limited to efficacy, i.e. PSA responses, pain responses, and objective responses and data on survival benefit are lacking, such rechallenge has become a less likely choice following the introduction of the new agents such as cabazitaxel, abiraterone, and enzalutamide, which have all demonstrated survival benefit in patients relapsing after docetaxel chemotherapy [28–30].
An alternative approach to the standard of 10–12 cycles docetaxel as used in the pivotal phase III studies is intermittent dosing of docetaxel suspending treatment after six cycles or at a predefined PSA decrease, and retreatment when PSA starts to rise again. In one of the larger studies a majority of patients responded again to such retreatment [31]. These data are of particular interest because of the absence of a defined optimal duration of chemotherapy in responding cases with mCRPC [26, 32]. In a prospective phase II study, patients were enrolled who had responded to first-line docetaxel and progressed after a chemotherapy-free interval of at least 5 months. Median overall survival since enrollment was 13 months, and a 50 % PSA decline was observed in 24.5 % of patients [25]. Like for docetaxel retreatment, OS data for intermittent docetaxel therapy are also lacking, and a second series of docetaxel has become questionable due to the newly available systemic treatment options.
Docetaxel-Based Combination Therapies
In the light of improved survival and modest toxicity with docetaxel as was demonstrated in TAX 327, numerous investigators, collaborative groups, and industry have investigated whether the efficacy of docetaxel could be improved by adding a second agent [33]. Here we will discuss docetaxel combination studies. An overview of the phase III combination trials with docetaxel is shown in Table 8.1.
Table 8.1
Phase III trials of docetaxel-based combinations
Agent | Result |
|---|---|
Docetaxel + GVAX (VITAL-2) | OS inferior in combination arm: 12.2 vs. 14.1 months HR 1.70 (95 % CI 1.15–2.53) |
Docetaxel + Calcitriol (ASCENT-2) | OS inferior in combination arm: 17.8 vs. 20.2 months HR 1.42 (95 % CI 1.13–1.86) |
Docetaxel + Atrasentan (SWOG S0421) | OS not improved in combination arm: 18 vs. 17 months HR 1.01 (95 % CI 0.87–1.18) |
Docetaxel + Zibotentan (ENTHUSE M1C) | OS not improved in combination arm: 20 vs. 19.2 months HR 1.00 (95 % CI 0.84–1.18) |
Docetaxel + Lenalidomide (MAINSAIL) | OS inferior in combination arm: 17.7 vs. median not reached HR 1.53 (95 % CI 1.17–2.00) |
Docetaxel + Bevacizumab (CALBG 90401) | OS not improved in combination arm: 22.6 vs. 21.5 months HR 0.91 (95 % CI 0.78–1.05) |
Docetaxel + Aflibercept (VENICE) | OS not improved in combination arm: 22.1 vs. 21.2 months HR 0.94 (95.6 % CI 0.82–1.08) |
Docetaxel + Dasatinib (READY) | OS not improved in combination arm: 21.5 vs. 21.2 months HR 0.99 (95 % CI 0.87–1.13) |
Docetaxel + Custirsen (SYNERGY) | Ongoing |
Phase III Trials
Immunotherapy
The GVAX platform of immunotherapies involved injection of cells derived from prostate cancer cell lines to provoke an immune response to multiple antigens expressed by the tumor cell. In addition, the cells were modified to secrete granulocyte macrophage colony-stimulating factor (GM-CSF). The VITAL-2 trial compared GVAX plus 3-weekly docetaxel with docetaxel plus prednisone and was interrupted early due to an unexpected higher death rate in the GVAX arm (67 deaths for GVAX plus docetaxel vs. 47 deaths for docetaxel plus prednisone) [34]. Another trial (VITAL-1) compared GVAX with docetaxel in patients with asymptomatic CRPC [35]. The study was prematurely terminated based on the results of a futility analysis conducted by the study’s Independent Data Monitoring Committee (IDMC) which determined that the study had less than a 30 % chance of meeting its predefined primary endpoint of improvement in overall survival.
Calcitriol
Calcitriol is an activated vitamin D analog that has shown to enhance antitumor activity of paclitaxel and docetaxel in vitro and in vivo [36, 37]. ASCENT-1 was a double-blind randomized phase II study that investigated weekly docetaxel plus high-dose calcitriol versus docetaxel plus placebo [38]. The primary endpoint PSA response rate did not differ between the treatment groups. Although it was not the primary endpoint of the trial, there was an improvement in OS for calcitriol over the placebo group. The ASCENT-2 trial was a randomized phase III trial designed to validate the observed survival benefit obtained with docetaxel plus calcitriol in the ASCENT trial [39]. In the phase III trial the control arm comprised the standard docetaxel regimen every 3 weeks. At an interim analysis, more deaths were noted in the ASCENT arm and consequently the trial was terminated early. Median OS was 17.8 months (95 % CI 16.0–19.5) for docetaxel plus calcitriol compared to 20.2 months (95 % CI 18.8–23.0) for docetaxel plus prednisone. Reasons for the worse OS by docetaxel plus calcitriol arm may have been attributed to the use of the weekly docetaxel schedule in the investigational arm [39].
Endothelin-A Receptor Antagonists
Atrasentan is an endothelin-A receptor antagonist that enhanced the effects of docetaxel against prostate cancer cells in vitro and in vivo [40, 41]. In the SWOG S0421 trial atrasentan plus docetaxel and prednisone was investigated in 991 patients with bone metastases. No difference in OS and PFS was observed for atrasentan plus docetaxel and prednisone compared with docetaxel and prednisone alone [42].
Another endothelin-A receptor antagonist zibotentan was investigated in the phase III trial ENTHUSE M1C combined with standard docetaxel versus docetaxel plus placebo. Docetaxel plus zibotentan did not result in a significant improvement in OS compared with docetaxel plus placebo (HR 1.00, 95 % CI 0.84–1.18) [43].
Angiogenesis Inhibitors
The oral angiogenesis inhibitor thalidomide demonstrated additive effects to taxane chemotherapy in vitro [44]. In two randomized phase II trials, the addition of thalidomide to docetaxel resulted in an encouraging PSA decline rates. Although more thromboembolic events were observed in patients treated with thalidomide, the combination regimen was reported to be well tolerated after the administration of prophylactic low-molecular-weight heparin [45, 46]. Lenalidomide is the successor of thalidomide with greater anti-angiogenesis efficacy as well as immunomodulatory effects. The randomized phase III trial (MAINSAIL) evaluated the efficacy of lenalidomide plus docetaxel versus docetaxel and placebo as first-line treatment for mCRPC. Following an interim analysis the study was stopped due to greater toxicity in the investigational arm and possibly reduced effectiveness. This could have been due to more frequent docetaxel dose reductions in patients allocated to lenalidomide [47].
Bevacizumab is a humanized immunoglobulin G monoclonal antibody to all the isoforms of VEGF-A. The CALBG Group investigated the addition of bevacizumab to standard docetaxel and prednisone in a randomized phase III trial. Despite an improvement in PFS and objective response, the addition of bevacizumab to docetaxel and prednisone did not improve OS and was associated with greater toxicity [48].
Aflibercept, a recombinant human fusion protein that binds A and B isoforms of VEGF and placental growth factor thereby inhibiting angiogenesis, was investigated in the phase III VENICE trial. In this study 1,224 men were treated with docetaxel plus prednisone and randomized to receive aflibercept or placebo. Median overall survival was 22.1 months (95.6 % CI 20.3–24.1) in the aflibercept group and 21.2 months (95.6 % CI 19.6–23.8) in the placebo group (stratified hazard ratio 0.94; 95.6 % CI 0.82–1.08; p = 0.38). The combination of aflibercept and docetaxel was associated with a higher incidence of grade 3/4 gastrointestinal disorders, hemorrhagic events, hypertension, fatigue, infections, and treatment-related fatal adverse events [49].
Bone Microenvironment Agents
SRC-family kinases play an important role in prostate cancer growth and invasion, as well as the pathogenesis of bone metastases and the regulation of osteoclast function [50–52]. Among others, dasatinib potently inhibits the SRC-family kinases (SRC, LCK, HCK, FYN, YES, FGR, BLK, LYN, and FRK [53]). In preclinical studies the tyrosine kinase inhibitor dasatinib inhibited cell duplication, migration, and invasion, and triggered apoptosis of tumoral cells. Dasatinib also acts on the tumor microenvironment, which is particularly important in the bone, where it inhibits osteoclastic activity and favors osteogenesis, exerting a bone-protecting effect [53]. These preclinical studies led to the hypothesis that combining dasatinib with docetaxel would improve treatment outcomes by targeting both the tumor and bone microenvironment. In a phase I/II study combining docetaxel with dasatinib, 18 out of 30 patients with measurable disease had a partial response and 14 patients had disappearance of lesions on bone scans [54]. However, the phase III READY trial demonstrated no survival benefit for docetaxel plus dasatinib compared to docetaxel and placebo [55].
Related posts:
 Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
 Phase I–II Targeted Treatments
Phase I–II Targeted Treatments
 Undifferentiated Prostate Cancer and the Neuroendocrine Phenotype
Undifferentiated Prostate Cancer and the Neuroendocrine Phenotype
 Epigenetics in Castration Resistant Prostate Cancer
Epigenetics in Castration Resistant Prostate Cancer
 Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


