Fig. 44.1
Partial karyotypes of chromosome abnormalities observed in lymphoid malignancies. The abnormal chromosomes are situated on the right side of each pair with the normal homologue on the left. (a) t(3;14)(q27;q32) involving the BCL6 gene on 3q27 and IGH@ gene on 14q32; (b) t(8;14)(q24;q32) involving the MYC gene on 8q24 and IGH@ gene; (c) t(11;14)(q13;q32) involving the CCND1 gene on 11q13 and IGH; (d) inversion of chromosome 14 involving the TRA@/D@ locus on 14q11.2 and the TCL1 gene on 14q32; (e) t(14;18)(q32;q21) involving the IGH@ gene and the BCL2 gene on 18q21
Mature B-Cell Neoplasms
Chronic Lymphocytic Leukaemia/Small Lymphocytic Lymphoma
Chronic lymphocytic leukaemia (CLL), or its non-leukaemic counterpart small lymphocytic lymphoma (SLL), is a chronic B-cell lymphoid neoplasm. Cytogenetic studies in CLL have been complicated by the particular difficulty in inducing the malignant cells to divide in culture. Early studies reported normal karyotypes for most cases of CLL, almost certainly because the metaphase spreads obtained in these cases originated from normal cells present in the blood or bone marrow cultures. The addition of B-cell mitogens such as 12-O-tetradecanoylphorbol-13-acetate (TPA) enabled the detection of cytogenetic abnormalities in approximately 35–45 % of CLL cases. Abnormalities of 11q and 13q were shown to be the most common findings in classical CLL [2] and trisomy 12 to be associated with atypical morphology [2, 3].
In 2006, Dicker and colleagues published a method of culturing CLL/SLL cells using a mixture of CpG oligonucleotide and interleukin-2, enabling abnormal karyotypes to be identified in more than 80 % CLLs by conventional cytogenetics [4]. The ability to visualise the karyotype of most CLL cases has confirmed that CLL is characterised by genomic imbalances, with the most common addition being trisomy 12 and the most common deletions being loss of 13q14 and 11q22 [5]. It has also identified a subgroup of patients with a complex aberrant karyotype (≥3 clonal chromosome aberrations) that are more likely to show deletion of 17p, an unmutated IGHV status and CD38 expression [5].
FISH applied to interphase cells has shown genetic abnormalities in most cases studied. A FISH study of 325 CLL cases identified abnormalities in 82 % cases, with deletions of 13q14 (55 %) and 11q22 (18 %), trisomy 12 (16 %) and deletions of 17p13 (7 %) and 6q (6 %) being the commonest aberrations found [6]. None of these aberrations was specific for CLL and so did not provide diagnostic information. However, FISH results correlated with outcome in this patient cohort. Patients with del(17p) and del(11q) had more advanced disease. Moreover, patients with del(17p) had the shortest median survival and those with del(13q) the longest. This and other studies have shown deletions of 17p13 and 11q22 to be prognostically significant markers in CLL [6–8]. Deletions of 17p13 have also been shown to correlate with the presence of TP53 mutations, with complex cytogenetic abnormalities [9] and with amplification of 2p and deletion of 6q [10]. Del(17p) may be acquired as a secondary event during the course of the disease, particularly in patients with unmutated IGHV genes.
The proportion of cells with a 17p13 deletion may also be an important prognostic factor. The UKCLL4 trial indicated that 17p deletion in >20 % of cells was associated with a dismal complete response rate and a 100 % progression rate within 3 years [11], but other groups have shown prognostic significance of 17p deletion using cut-off values of <20 % based on the level of aberrant signal patterns identified in normal controls [12, 13].
FISH studies of 18 patients followed for more than 10 years and never requiring treatment showed that none of them had deletions of 17p (TP53) or 11q (ATM), two had trisomy 12 and three had a deletion of 13q. In 13 patients, none of the studied abnormalities was detected. Thus, in a group of patients with demonstrated indolent disease, the absence of genomic aberrations as demonstrated by FISH correlated with good prognosis [14].
Loss of 13q14, as detected by FISH using a D13S319 probe, is the commonest finding by FISH. The focus of the deletion has been identified as two miRNAs, MIR15A and MIR16-1 located in the fourth intron of the DLEU2 gene [15]. It has been noted that both alleles may be lost and one study of 30 patients suggested that loss of both copies of 13q14 might be a marker for more aggressive disease [16]. Van Dyke et al. grouped 323 patients with a deletion of 13q as a sole abnormality according to locus dosage. Time to first treatment and survival were equivalent for patients who were heterozygous, homozygous or mosaic for 13q deletion, suggesting that it is the presence or absence of a deletion rather than the mono- or bi-allelic nature of the deletion that dictates clinical behaviour [17]. Accurate mapping of 13q14 deletions using oligonucleotide DNA array technology has enabled small type I lesions (without RB1 gene deletion) to be distinguished readily from larger type II deletions (where RB1 is deleted in addition to the MIR16/MIR15A cluster) [18]. At this stage, the clinical significance of this information remains to be determined.
The presence of translocations, either balanced or more commonly unbalanced, appears to indicate a poor prognosis. Translocations have been associated with significantly shorter treatment-free survival (TFS). Moreover, the favourable effect of del(13q) as a sole abnormality detected by FISH is undermined by the presence of translocations, leading to a significantly shorter TFS [19]. Apparently balanced reciprocal translocations involving 13q14 have been observed in CLL, but FISH has shown that more than 90 % demonstrated a 13q14 deletion at the translocation breakpoint [5]. Patients with del(11q) detected by FISH plus translocations also showed a significantly shorter TFS when compared with patients with del(11q) without translocations. Most cases of del(17p) detected by FISH were associated with the presence of translocations [19, 20]. A recurrent dicentric unbalanced 17;18 translocation was observed in 16/1213 (1.3 %) CLL cases, resulting in the loss of much of 17p and 18p and associated with early age at diagnosis and accelerated disease progression [21].
Reciprocal translocations are uncommon in CLL [5]. IGH@ translocations have been reported in 5 %–15 % cases [5, 22–24]. FISH studies have shown that the presence of an IGH@ translocation predicts a significantly shorter overall survival [24]. The poor prognosis associated specifically with t(14;18) in CLL has been disputed by Put et al., who reported 40 patients with CLL and t(14;18) or a variant translocation. The median TFS in this cohort was 48 months, the estimated median overall survival 182 months from diagnosis and the t(14;18) was not associated with advanced disease [25]. However, the adverse impact of the t(14;18) has been confirmed by others [22].
Another IGH@ translocation, t(14;19)(q32;q13), has been observed in a variety of B-cell lymphoid malignancies, including CLL, marginal zone lymphoma (MZL) and lymphoplasmacytic lymphoma (LPL). The translocation results in the BCL3 gene on 19q13 being juxtaposed to the IGH@ gene on 14q32, resulting in overexpression of BCL3 [26]. The t(14;19) in CLL has been most commonly associated with atypical morphology and immunophenotype, a younger age group with a high proportion of patients diagnosed at less than 40 years of age, the presence of trisomy 12 and deletion of 6q, unmutated IGHV genes, rapidly progressive disease and a poor prognosis [26, 27].
Another rare but recurrent IGH@ translocation in CLL is the t(2;14)(p16;q32), juxtaposing the BCL11A gene on 2p16 and the IGH@ gene on 14q32. CLL with t(2;14) is associated with atypical morphology and unmutated IGHV genes [28]. MYC translocations are rare in CLL but have been reported in association with increased prolymphocytes, complex cytogenetic abnormalities and a poor prognosis [29].
Interstitial deletions of 14q are a recurrent abnormality in CLL. The breakpoints of the del(14)(q24.1q32.33) have been mapped to within the IGH@ locus and in a region of 14q24.1 containing the ZFP36L1 gene. One gene located within the deleted region is the candidate tumour suppressor gene (TSG) TRAF3 which has been shown to be homozygously deleted in CLL, or heterozygously deleted with a loss of function mutation in the remaining allele and associated lower expression of TRAF3 mRNA [30]. Deletion of TRAF3 is one of many genetic events predicted to perturb NFKB signalling in mature B-cell lymphomas.
FISH studies have also identified patients with deletions of IGH@. In a series of 291 CLL cases, FISH identified loss of the telomeric 5′ segment of IGH@ (using a 900-kb probe covering the entire variable IGH segment) in 82 % cases and found the deletion to be associated with good prognosis markers, including del(13q), normal karyotype and lack of CD38 and ZAP-70 expression [23]. However, it appears that these V H deletions are not oncogenic but rather reflect the physiological events accompanying somatic V–D–J assembly [31].
Genetic factors appear capable of predicting response to therapy. The response to rituximab therapy is significantly lower in CLL patients with del(17)(p13) when compared to that in patients with other genetic abnormalities identified by FISH [32], possibly due in part to significantly lower CD20 expression [33]. However, the adverse impact of 11q22 and 17p13 deletions may be overcome by combinations of chemotherapy and immunotherapy, such as alemtuzumab [34, 35]. A prognostic scoring system for CLL has been proposed that incorporates age (≥65 years), white blood cell count (≥20 × 109/L), unmutated IGHV status, TP53 deletion, the presence of an IGH@ translocation and the number of chromosome aberrations identified by conventional cytogenetics [24]. Using these parameters, patients could be classified into favourable, intermediate and unfavourable risk groups.
SNP genomic arrays have revealed the common abnormalities normally identified by FISH (trisomy 12, deletion of TP53, deletion of ATM and deletion of 13q14) in the majority of CLL samples [36]. In addition to the common abnormalities, numerous other copy number changes and uniparental disomy (UPD) have been detected. UPD involving the entire length of chromosome 13 and resulting in homozygous deletion of MIR15A and MIR16-1 in the minimally deleted region of 13q14 has been reported [36], together with gain of MYC at 8q24, deletion of 8p (TRAIL1/2) and gain of 2p [37]. The region of gain of 2p in 8 %–28 % of CLL cases and up to 39 % of CLLs with del(17p) includes the oncogenes, MYCN, BCL11A and REL [38]. Genomic complexity, as measured by the number of copy number changes identified by SNP array platforms, has emerged as an independent risk factor with increased complexity correlating with shortened TFS and time to subsequent therapy [39], possibly due in part to the association of del(17p) with higher numbers of copy number aberrations [10, 40]. Array CGH of del(17p) CLLs showed deletions of 18p and/or 20p to be exclusively associated with TP53 deletion, and the higher the percentage of TP53 deleted cells, the greater the number of additional cytogenetic abnormalities [10]. Array CGH narrowed regions involved in recurrent abnormalities to an 18.8-Mb deleted region of 11q, encompassing ATM, a deletion of 13q encompassing MIR15A/MIR16-1 and a gain of 2p16 (100 kb) covering the REL and BCL11A genes [41].
Finally, given the median age of onset of CLL/SLL, the question must be asked as to whether it is cost effective and likely to impact on the choice of therapies to apply a wide battery of tests to older patients. Shanafelt et al. divided a cohort of 2,487 CLL patients into those aged <55, 55–64, 65–74 and ≥75 years at diagnosis and determined that those aged ≥75 years did not show a reduced life expectancy when compared with age-matched controls and that prognostic testing in this age group had little utility for predicting overall survival [42].
B-Cell Prolymphocytic Leukaemia
B-cell prolymphocytic leukaemia (B-PLL) is a rare neoplasm of B prolymphocytes; it is differentiated from CLL by prolymphocytes representing 55 % or more of lymphoid cells in the peripheral blood. Few studies of the cytogenetics of B-PLL have been reported and a retrospective analysis of eight cases of B-PLL with t(11;14) suggested that these cases represented a splenomegalic form of mantle cell lymphoma (MCL) evolving to leukaemia [43]. Whilst trisomy 12 appears rare, deletions and mutations of TP53 and deletions of 11q22.3–q23.1 and 13q14 have been observed in 53 %, 39 % and 27–55 % of cases respectively [44, 45]. The frequency of TP53 deletions and mutations may explain the aggressive course usually seen in B-PLL. The Burkitt 8;14 translocation and variant 8;22 and 2;8 translocations have been reported in a handful of cases of B-PLL [46–48]. Gene expression analysis comparing B-PLL with CLL showed the two disorders to be biologically distinct; one of the genes significantly over-expressed in B-PLL in comparison with CLL was MYC [49].
Splenic Marginal Zone Lymphoma
Splenic marginal zone lymphoma (SMZL) is an uncommon low-grade small B-cell disorder which usually presents with a CD5 negative lymphocytosis. Complex chromosomal aberrations are seen in more than 50 % cases.
The commonest abnormalities are gains of 3/3q (15–32 %), trisomy 18 (8–12 %), trisomy 12 (5–20 %) and deletion of 7q32 (21–44 %) [50–52]. The deletion breakpoints at 7q appear heterogeneous with the common area of loss localised using aCGH to 7q32.1–7q32.2 [53]. The presence of two or more chromosomal aberrations may correlate with a shorter survival, but the previous reports of an adverse outcome in patients with del(7q) do not appear to be substantiated in larger series [54, 55]. The smallest overlapping region of gain of chromosome 3 was narrowed down to 3q26.33q29 [41].
Immunoglobulin (IGH@, IGK@, IGL@) translocations have been observed in 7 %–21 % SMZL with a variety of partner chromosomes, resulting in translocations including t(9;14)(p13;q32) (IGH@-PAX5), t(3;14)(q27;q32) (IGH@-BCL6), t(14;19)(q32;p13) (IGH@-IRF4/MUM1), t(14;19)(q32;q13) (IGH@-BCL3), t(6;14)(p21;q32) (IGH@-CCND3) and t(2;7)(p11;q21–22) (IGK@-CDK6). Numerous other IG translocations have been observed, but the partner genes have not been identified [55, 56]. Translocations involving BCL2 and MALT1 have not been seen in SMZL and although the t(11;14) that is considered characteristic of MCL has been described in cases of SMZL, these were reclassified subsequently as MCL. Most cases of SMZL with IG translocations have been shown to contain complex cytogenetic abnormalities in addition to the translocation, but del(7q) and IG translocations rarely coexist [52].
Hairy Cell Leukaemia
Hairy cell leukaemia (HCL) has proven difficult to analyse cytogenetically as the bone marrow aspirate is frequently unsuccessful due to marrow fibrosis. A study using IL4 and anti-CD40 monoclonal antibodies to induce proliferation was able to successfully karyotype 42/43 patients tested, but the abnormality rate was only 19 % with trisomy of chromosome 5, abnormalities of chromosome 7 and structural rearrangements of 14q being the most frequent structural and numerical abnormalities [57].
A study of splenic tissue from 14 patients with HCL, using CGH and FISH, identified chromosomal gains and losses in only 4/14 cases [58], and a conventional cytogenetics study by Sambani et al. also only found an abnormal karyotype in 8/18 evaluable patients, with the commonest abnormality identified being rearrangements of 14q [59]. Further aCGH and SNP arrays also reported few recurrent abnormalities but identified trisomy 5 and abnormalities of 14q22–32 in a minority of cases [60]. Nuclear expression of SOX11 has been observed in a subset of HCL associated with up-regulation of cyclin D1, but these cases did not contain the t(11;14) that is responsible for the over-expression of cyclin D1 in MCL [61].
Lymphoplasmacytic Lymphoma
LPL is a malignant B-cell lymphoma characterised by proliferation of pleomorphic lymphoplasmacytoid lymphocytes and monoclonal IgM production. Cytogenetic studies of LPL have shown the most common chromosome abnormalities to be deletion of the long arm of chromosome 6, del(6q), observed in 7 %–55 % of cases, and trisomy 4; IGH@ rearrangements are rarely observed [62]. However, FISH studies found the del(6q) to be an uncommon finding in patients with nodal LPL [63, 64]. Trisomy 3 or 3q, resulting from an isochromosome of 3q, has also been associated with B-cell chronic lymphoproliferative disorders with plasmacytoid differentiation [65, 66]. Array CGH applied to LPL cases has shown del(6q), loss of chromosome 17, gain of 3q, gain of chromosome 7 and monosomy 19 in some but not all cases [67].
The t(9;14)(p13;q32) involving the PAX5 and IGH@ genes has been reported in a subset of nodal-based small cell lymphomas with plasmacytoid differentiation [68], but has not been observed in other series of patients with Waldenstrom’s macroglobulinaemia [69–71]. Moreover, the t(9;14) has been observed in a number of other lymphoma subtypes, including CLL, T-cell-rich B-cell lymphomas and DLBCL [72].
To make the diagnosis of LPL, other B-cell lymphoid neoplasms that may show some plasmacytoid differentiation such as CLL/SLL, follicular lymphoma or MZL must be excluded. Differentiating between LPL and mucosa-associated lymphoid tissue (MALT) lymphoma, in particular, may be problematic but LPL has not been shown to harbour the t(11;18), t(1;14) or t(14;18) translocations that are frequently observed in MALT lymphomas [73, 74].
Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma)
The MALT lymphomas are common low-grade B-cell lymphomas that arise on a background of chronic inflammation at various mucosal sites. The gastric MALT lymphomas are linked to Helicobacter pylori (H. pylori) infection, and small intestine, skin and ocular adnexal disease have been linked to Campylobacter jejuni, Borrelia burgdorderi and Chlamydia psittaci infections respectively. MALT lymphomas are characterised by trisomies of chromosomes 3, 12 and/or 18 and a series of reciprocal translocations: t(11;18)(q21;q21), t(1;14)(p22;q32), t(14;18)(q32;q21) and t(3;14)(p14.1;q32) involving the API2-MALT1, IGH@-BCL10, IGH@-MALT1 and IGH@-FOXP1 genes respectively. The incidence of these translocations varies according to the anatomical site of the lymphoma (Table 44.1). The t(11;18) is seen predominantly in stomach and lung; the t(14;18) in liver, ocular adnexa/orbit, skin and salivary glands and the trisomies in intestinal and salivary gland lymphomas [75, 76]. The oncogenic products of the t(11;18)(q21;q21), t(1;14)(p22;q32) and t(14;18)(q32;q21) all target the NFKB pathway, which is considered critical to malignant B-cell transformation and lymphoma progression.
Table 44.2
Additional abnormalities in mantle cell lymphoma
Common abnormalities | Recurrent breakpoints | References |
|---|---|---|
+Xq | [147] | |
–Y | ||
del(1p) | 1p31–32, 1p21–22, 1p36 | [138] |
+3 | [138] | |
+3q | 3q27–28 (BCL6) | |
+4p | 4p12–13 | [427] |
del(6q) | 6q21 6q27 | |
del(7q) | 7q22–35 | [143] |
del(8p) | 8p22, 8p11, 8p21.3 (TRAIL-R1 and TRAIL-R2) | [132] |
+8q24 | 8q24 (MYC) | [132] |
–9 | [138] | |
del(9p) | 9p21 (INK4A/ARF) | |
del(9q) | 9q21 | [138] |
+10p | 10p15 | [132] |
del(10q) | [142] | |
+11q | 11q13 (CCND1) | [428] |
del(11q) | 11q22.2 (ATM), 11q23.3 | |
+12 | [138] | |
+12q | 12q12–13 | |
–13 | [138] | |
del(13q) | 13q13–14 13q21–32 13qter | |
+13q | 13q31.3 (MIR17HG) | [428] |
del(15q) | 15q23 | [132] |
+15q | 15q23 | [132] |
del(17p) | 17p13.1 (TP53), 17p13.3 | [132] |
–18 | [142] | |
+18q21 | [132] | |
+19p13.3 | [143] | |
del(20p) | 20p12.1–12.3 | [427] |
del(20q) | 20q12–q13.2 | [427] |
del(22q) | 22q12.1–12.3, 22q13.31–13.32 | [427] |
In the t(11;18), the apoptosis inhibitor gene AP12 at 11q21 and MALT1 at 18q21 cause a hybrid gene AP12/MALT1 to form on the derivative chromosome 11. Occasional variant translocations involving 11, 18 and a third chromosome have been described, all resulting in the AP12/MALT1 fusion [77]. MALT lymphomas are known to transform to DLBCL, but the t(11;18) has only rarely been identified in DLBCL [78]. It appears that the t(11;18) negative lymphomas develop secondary aberrations and are the only MALT lymphomas capable of transformation to DLBCL [79].
A second translocation involving the MALT1 gene at 18q21 is the t(14;18)(q32;q21), which has an identical conventional cytogenetic appearance to the t(14;18) characteristically seen in follicular lymphoma (FL). The t(14;18) in MALT lymphomas juxtaposes MALT1 at 18q21, rather than BCL2, adjacent to the promoter region of the IGH@ gene on 14q32, resulting in deregulation of MALT1 expression [75, 80]. Trisomy 3 and/or trisomy 18 has been observed in association with the t(14;18). Lymphomas with t(14;18) show strong cytoplasmic expression of both MALT1 and BCL10 [81]. Unlike the indolent MALT lymphomas that are dependent on the continuous presence of H. pylori, MALT lymphomas with translocations targeting the MALT1 gene can be found in cases without H. pylori and are resistant to H. pylori therapy.
Rare cases of t(14;18) involving the BCL2 gene with IGH@ rather than MALT1 have been described in MALT lymphomas. Although it has been suggested that these cases cluster in Hepatitis C-positive patients [82], a case of IGH@-BCL2-positive MALT lymphoma in a Hepatitis C-negative patient who responded to H. pylori eradication therapy has been reported [83].
A less common translocation observed in MALT lymphomas is the t(1;14)(p22;q32) which transfers the entire BCL10 gene to chromosome 14 where its expression is inappropriately stimulated by the neighbouring IGH enhancer. The upregulated BCL10 gene product is truncated and loses its weak pro-apoptotic function but retains its ability to activate NFKB. Nuclear BCL10 expression is also observed frequently in MALT lymphomas that do not exhibit the t(1;14) and has been also associated with the presence of the AP12-MALT1 fusion protein [84]. Rare cases of t(1;2)(p22;p12) have also been reported juxtaposing BCL10 adjacent to the immunoglobulin kappa (IGK@) light chain gene [85].
Another translocation involving the IGH@ gene on chromosome 14, t(3;14)(p14.1;q32) has been described in MALT lymphomas, but is also reported in DLBCL and MALT lymphomas with a DLBCL component [86, 87]. The translocation involves the partner gene FOXP1 on chromosome 3. Trisomy 3 has been observed in conjunction with the t(3;14) and three copies of the FOXP1 gene were identified in 17 % MALT lymphomas and 12 % MALT lymphomas with a DLBCL component. However, FOXP1 expression appears independent of the t(3;14) translocation or of copy number changes of FOXP1. The t(3;14) does not occur in association with any of the other three primary translocations characteristic of MALT lymphoma [86]. Other rare IGH@ translocations have been identified in single cases of MALT lymphoma, involving the partner genes CNN3 (1p), ODZ2 (5q) and JMJD2C (9p) [88].
The three translocations all appeared to occur mutually exclusively in a series of 252 primary MALT lymphomas, with the t(11;18), t(14;18), t(1;14) and trisomies of 3 and/or 18 seen in 13.5 %, 10.8 %, 1.6 % and 42.1 % of cases respectively [76], and the t(11;18) and trisomies of 3 and/or 12 appear unlikely to occur together [89].
Using FISH, trisomies of 3, 12 and 18 were frequently observed in gastric, parotid and thyroid MALT lymphomas but not in most cases of primary breast MALT lymphomas. MALT1 gene rearrangements are also rare in breast MALT lymphomas [90]. Trisomies of 3 and 18 have also been observed in a high proportion of primary intestinal MALT lymphomas [91]. A recurrent deletion of a 2.9-Mb region at 6q23.2 to 6q24.1 has been observed particularly in ocular adnexal MALT lymphomas, and the target gene for the deletion is thought to be TNFAIP3, an inhibitor of NFKB signalling [92].
Detection of the common translocations may be via conventional cytogenetics, FISH or RT-PCR. FISH has been applied to cytogenetic suspensions and also to paraffin-embedded tissue. Remstein et al. used two-colour FISH to identify the t(11;18) in nuclei isolated from paraffin-embedded tissue in MALT lymphomas in which the t(11;18) had been previously identified by RT-PCR [89]. FISH showed a fusion signal for the t(11;18) in 92 % of RT-PCR-positive cases. This study also confirmed that t(11;18) was rarely observed with trisomy 3 or 18.
Eradication of H. pylori infection with antibiotics induces complete remission in 70–80 % MALT lymphomas but, for gastric MALT lymphomas, the t(11;18) appears to be a clonal marker for resistance to H. pylori eradication therapy and antigen-independent growth. Liu et al. studied a series of cases with H. pylori gastritis, MALT lymphomas and mucosal DLBCL [93]. The AP12/MALT1 fusion transcript was not detected in H. pylori gastritis and mucosal DLBCL but was found in 25/72 (35 %) of MALT lymphomas at various sites. Its incidence was significantly higher in gastric cases that showed dissemination to lymph nodes or distal sites compared with those confined to the stomach.
Lui et al. extended their studies to 111 patients with H. pylori-positive gastric MALT lymphoma who were treated with antibiotics [94]. The t(11;18) was detected in 2/48 complete-regression cases, both of which relapsed subsequently without H. pylori reinfection. The translocation was found in 42/63 non-responsive cases, including 26/43 stage 1E. Its presence therefore appeared to be a strong indicator for non-response to antibiotics. Although there have been cases of t(11;18)-positive gastric MALT lymphoma that have achieved durable remissions with H. pylori eradication therapy alone [95], the t(11;18) has also been shown to be predictive of resistance to oral alkylating agents with less than 10 % durable remission at long-term follow-up [96].
BCL10 nuclear expression is observed in MALT lymphomas in the presence of the t(1;14) which results in upregulation of BCL10 but is also observed in lymphomas in the absence of translocations that directly target BCL10. Normal B cells express BCL10 in the cytoplasm but MALT lymphoma cells carrying either t(1;14) or t(11;18) show nuclear expression. Such expression has been correlated with advanced disease [93] and resistance to H. pylori eradication therapy [97, 98].
Ferreira et al. studied 9 MALT lymphomas by aCGH and demonstrated an altered genome in only 33 %, with no recurrent overlapping regions of imbalance identified [41]. Array CGH of 19 salivary gland MALT lymphomas identified recurrent gains of 1p32-1pter in 43 %, 9q33–34 in 84 %, 11q11–13 in 42 %, chromosome 17 in 58 % and 18q21–22 in 42 %. Gains of 9q34, 11q13 and 18q21 were frequently concurrent [99]. Deletion of 6q23.3 containing the NFKB inhibitor A20 was observed in MALT lymphomas of the ocular adnexa, salivary gland, thyroid and liver but not lung, stomach or skin. In ocular cases, A20 deletion was associated with a higher proportion of relapse and a shorter relapse-free survival [100].
Array CGH was also used to compare MALT lymphomas with and without the t(11;18). Chromosome gains and losses were considerably more frequent in t(11;18)-negative lymphomas, with gains more frequent than losses; these included all or part of chromosomes 3, 12, 18 and 22 in 23 %, 19 %, 19 % and 27 % respectively, and gain at 9q34, the site of the TRAF2 and CARD9 genes (interaction partners of BCL10 in NFKB activation) in 42 % [101]. In those cases without t(11;18), resistance to H. pylori therapy was marked by a greater number of genomic imbalances, particularly gain of 1p36.2, chromosome 3 and 18q and loss of 1p36.3 and 7q31–q33 [102]. Moreover, cases with trisomy 3 were H. pylori negative, t(11;18) negative and resistant to eradication therapy [102]. In t(11;18)-negative gastrointestinal MALT lymphomas, trisomy of 18q21, including MALT1, detected by FISH has been associated with an adverse outcome [103] and the presence of trisomy 18q21 has been strongly correlated with trisomy for 3q27/BCL6 [104].
Gene expression analysis has revealed that MALT lymphomas harbouring either t(11;18), t(1;14) or t(14;18) show enhanced expression of NFKB target genes, whilst translocation-negative cases show active inflammatory and immune responses [105].
Nodal Marginal Zone Lymphoma
Trisomy 3 is recognised as the commonest recurrent cytogenetic finding in nodal MZL. Other frequently observed abnormalities include trisomy 18 and structural abnormalities of chromosome 7 [106, 107]. A cytogenetic study of 103 MZL cases with an abnormal karyotype showed trisomy 3 or an extra copy of the long arm of chromosome 3 to be present in 38/103 (37 %) of cases and as a sole abnormality in the stemline of 5/38 cases, but with clonal evolution evident in sidelines [107]. The 3q24 to 3qter segment comprised the most consistently over-represented region. Chromosome 7q deletions were the second most common abnormality, observed in 32/103 patients and in 9 as a sole abnormality, occurring as either an interstitial deletion or an unbalanced translocation. The consistently deleted region was 7q33 to 7q35. Four patients had an unbalanced 3;7 translocation resulting in trisomy of 3q and a 7q deletion. Trisomy 18 or an extra copy of 18q was identified in 29/103 patients, 6 of whom showed trisomy of 18q as a result of an isochromosome of 18q. Deletion of 6q was seen in 20/103 patients, trisomy 12 in 15 patients and abnormalities of chromosome 8 leading to deletion of 8p in 15 patients, three of whom had an isochromosome of 8q. Lymphoma-associated translocations were observed in only 13 % patients, including four with the typical MCL t(11;14), four with t(3;14)(q27;q32) or variants, three with t(9;14)(p13;q32) and 2 with t(14;19)(q32;q13.1). Comparing their findings with karyotypes of patients with SLL/CLL, MCL and FL, the combination of +3 and del(7q), which was present in 10 % MZL, was not observed in any of the other three diagnoses. However, as trisomies of 3, 12 and 18 and deletions of 6q and 7q are all recognised abnormalities in lymphoproliferative disorders, they cannot be regarded as specific for MZL.
Follicular Lymphoma
FL represent approximately 20–40 % of non-Hodgkin lymphomas and are generally associated with an indolent clinical course and overall median survival of 8–10 years. The t(14;18)(q32;q21) translocation or its variants, t(2;18)(p12;q21) and t(18;22)(q21;q11.2), are observed in approximately 85 % of FL. FL cells are localised to the germinal centre. The characteristic t(14;18)(q32;q21) juxtaposes the IGH@ gene at 14q32 to the BCL2 gene on 18q21 (Fig. 44.1). This juxtaposition results in over-expression of the BCL2 protein, which has anti-apoptotic properties. The BCL2 protein is not normally expressed in germinal centre cells.
The variant translocations, t(2;18)(p12;q21) and t(18;22)(q21;q11), involve the IGK@ or IGL@ genes respectively rather than IGH@. There have also been complex translocations described involving a third chromosome breakpoint in addition to 14q32 and 18q21 [108]. The result of all of these rearrangements appears to be the same, over-expression of BCL2. Despite the anti-apoptotic activity of BCL2, low-grade follicular lymphoma cells carrying the t(14;18) die quickly by apoptosis when cultured in vitro, making cytogenetic studies difficult in this disorder.
The t(14;18) is observed in most grade 1 and grade 2 FL, but in fewer grade 3a and a minority of grade 3b cases. The t(14;18) is also less frequently observed in FL arising in extranodal sites such as skin or testis. Guo et al. [109] identified 69 % of FL as “typical” with low histological grade (1 and 2) co-expression of BCL2 and CD10 and containing the t(14;18). They considered this group to be a homogeneous disease entity but the rest to comprise a group of heterogeneous disorders. BCL2 expression may be demonstrated in some cases by immunohistochemistry in the absence of the t(14;18) and there is a case report of a FL with a t(2;18)(q11.2;q21) showing BCL2 over-expression as a result of juxtaposition to the AFF3 gene at 2q11.2, a non-IG translocation [110]. A comparison of gene expression profiles between t(14;18)-positive and negative FL showed germinal centre B-cell associated signatures in t(14;18)-positive cases, whilst activated B-cell like, NFKB proliferation and bystander cell signatures were observed in the t(14;18)-negative cases. In 32 % of t(14;18)-negative FL, there was weak or absent CD10 expression and most showed an increased Ki67 proliferation rate. Despite these distinct differences, no survival difference was observed between the two groups [111].
Another array CGH study found that trisomy 3, gain/amplification of 18q21-qter (including BCL2) and loss of 9p21 (CDKN2A locus) were significantly more frequent in t(14;18)-negative cases. Most of the t(14;18)-negative cases that showed BCL2 expression by immunohistochemistry exhibited gain/amplification of 18q21-qter, and the majority were CD10 negative and MUM1 positive, suggesting that they were derived from late germinal centre B cells [112].
Additional genetic aberrations are observed in more than 90 % of FL as, although the t(14;18) is considered the primary genetic event in FL, additional changes are required to produce the fully malignant phenotype. The most frequent secondary numerical aberrations include an extra X chromosome and trisomies of 7, 12 or 12q, 18, or a doubling of the der(18)t(14;18). Structural abnormalities frequently involve chromosomes 1, 6q and 10q22q24 [113–116]. There has been shown to be a correlation of del(1)(p36), del(6q), del(10q) and trisomy 7 with morphological progression but, beyond that, a consistent pattern has not been discerned. M-FISH analysis of a series of FL identified the commonly observed secondary abnormality add(1)(p36) to be an unbalanced 1;17 translocation in approximately one-third of cases [114].
Transformation to higher grade lymphoma occurs in 25 %–60 % of the cases and is frequently accompanied by the further acquisition of secondary genetic changes. An average number of six chromosomal abnormalities were reported in a FL karyotype in addition to the t(14;18) [113]; most of these changes were unbalanced, resulting in gain or loss of chromosomal material. The complexity of secondary abnormalities appears to correlate with grade; higher grade FL contain more abnormalities.
A microsatellite study of 9p21 markers in 11 cases of FL that had progressed to DLBCL identified 9p deletions in 73 % at transformation, suggesting that del(9p) might be an important secondary event in the histological progression of FL [117]. In another study of 210 patients with FL, 31 % of whom transformed to higher grade lymphoma over a 10-year period, an additional X chromosome in the diagnostic karyotype of male patients was the only predictor of adverse outcome [118].
Trisomy 7, deletion of 6q and an additional copy of the derivative 18, der(18)t(14;18) were identified as early additional genetic changes, with each indicating the first step in different “pathways” of evolution. Deletion of 17p and trisomy 12 were significant negative predictors of overall survival in one large series [119], and del(6q), +5, +19 and +20 were associated with poorer overall survival and del(17p) with poorer event-free survival in another [115]. Deletion at 1p36 was identified in 27/29 t(14;18)-negative FLs and was correlated with predominantly diffuse morphology, often with low clinical stage but large localised inguinal tumours [120].
Using aCGH, 16 recurrent regional aberrations were identified that were independent predictors of overall survival in multivariate analysis on the following chromosomes:1p-, 1q+, 5p+, 6q-, 8q+, 17p- and 17q+; two of these, deletions of 1p and 6q, were also shown to be predictors of transformation [121].
A subset of t(14;18)-negative FL contains a t(3;14)(q27;q32) translocation (Fig. 44.1), resulting in over-expression of BCL6 on 3q27. BCL6 rearrangements (3q27) are less common in FL than in DLBCL and are detected in 9–14 % of FL cases by conventional cytogenetics. Paradoxically, the highest levels of BCL6 expression are found in FL in cases that do not contain 3q27 rearrangements [122]. The t(14;18) is least commonly observed in FL 3b subtype, lymphomas composed almost exclusively of centroblasts. This group is most likely to contain a 3q27 rearrangement with secondary abnormalities more like DLBCL [123, 124]. Analysis of BCL6 aberrations by FISH in 100 t(14;18)-negative FLs showed that 77 contained a BCL6 gene rearrangement or translocation and/or gain or amplification of the BCL6 gene, whilst only 23 had no BCL6 gene abnormalities. The cases with BCL6 gene gain/amplification were more likely to show higher grade morphology, higher BCL2 and MUM1 expression and more likely to also show gain of 18q21 (BCL2) than other groups [125]. Jardin et al. studied 15 cases of FL with 3q27 translocations. Thirteen of the 15 had a t(3;14)(q27;q32), most were CD10 negative and BCL2 negative but there did not appear to be any significant differences in age, sex, performance status, bone marrow involvement or overall survival, when compared with FL with t(14;18) [122].
Balanced translocations, apart from the t(14;18) and t(3;14), are infrequent but usually represent lymphoma-specific changes when present, such as the Burkitt t(8;14) translocation or its variants associated with high-grade transformation. At transformation, 8q24 (MYC) translocations are common [118]. Of interest is the observation that the FL studied serially showed loss of some cytogenetic aberrations that had been present at diagnosis over time [126]. A small number of FL cases have been reported with a MYC translocation present at diagnosis in addition to the t(14;18); this finding may be associated with an aggressive clinical course [126]. However, Ladanyi et al. identified six cases with t(8;14), but no evidence of the t(14;18) in a series of 278 karyotypically abnormal FL. Sequencing showed that the breakpoints did not lie in the regions usually involved in the t(8;14) of Burkitt lymphoma. Moreover, the cases had an indolent course, suggesting that the t(8;14) in FL is not always the same translocation as that seen in high-grade lymphomas [127].
It is not generally considered necessary to identify the t(14;18) to make a diagnosis of FL, but it may be useful to confirm the diagnosis in small biopsies or with atypical morphology when other tests are inconclusive. Testing for t(14;18) can also be used to monitor therapy, detect recurrent disease and determine the effectiveness of bone marrow purging prior to autologous stem cell harvest. Conventional cytogenetic analysis can detect the t(14;18) but requires fresh tissue with viable dividing cells; in many centres, lymph node biopsies are not routinely submitted for karyotyping.
FISH can be applied to cytogenetic preparations and is also able to detect the t(14;18) in smears, cytospin preparations, touch imprints, frozen tissue and fixed paraffin-embedded tissue. It is best in the identification of the t(14;18) in diagnostic specimens and less effective in detecting residual disease. FISH has a higher sensitivity than PCR as FISH probes span almost all breakpoints, whereas standard PCR primer sets cover only a limited number of breakpoint regions. However, PCR analysis is more effective for the detection of minimal residual disease [128].
Mantle Cell Lymphoma
MCL, an aggressive lymphoid tumour, is characterised by distinct clinical, morphological and genetic features. Whilst morphology and immunophenotyping have been widely used to define this entity, the most accurate method of diagnosis is via genetic methods.
The cytogenetic hallmark is the translocation t(11;14)(q13;q32) (Fig. 44.1), which juxtaposes the cyclin D1 gene (CCND1, previously known as BCL1and PRAD1) with an enhancer sequence of the IGH@ gene at 14q32. Detection of the t(11;14) may be via conventional cytogenetics or FISH using an IGH@/CCND1 dual colour dual fusion translocation probe, and FISH may be used on peripheral blood, bone marrow or lymph node samples (Fig. 44.2). Successful FISH identification of the t(11;14) has also been reported on archived fine-needle aspirate cytology slides [129] and paraffin sections [130]. The finding of the t(11;14) is of diagnostic rather than prognostic significance in MCL. The translocation does not disrupt CCND1 but results in over-expression and a quantitative increase in normal cyclin D1 protein. Whilst almost half of the 11q13 breakpoints of the t(11;14) in MCL cluster in an 8–10-kb major translocation cluster region, the other 11q13 breakpoints are scattered over more than 100 kb, making PCR-based approaches to identifying the translocation problematic. Comparative analyses of interphase FISH for detection of the t(11;14) with other methods such as conventional cytogenetics, PCR for amplification of the t(11;14) genomic breakpoint, competitive RT-PCR for detection of cyclin D1 transcript over-expression and immunohistochemistry for cyclin D1 protein detection identified FISH as by far the most sensitive method for detecting evidence of the t(11;14) [130–132].
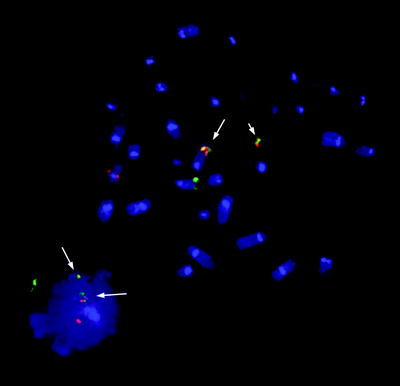
Fig. 44.2
Metaphase spread and interphase cell from a patient with mantle cell lymphoma showing IGH@-CCND1 fusion signals (arrowed). The two fusion signals shown on the metaphase spread are located on the der(11)t(11;14) (short arrow) and on the der(14)t(11;14) (long arrow). The SpectrumOrange™ signal (CCND1) is on the normal chromosome 11 and the SpectrumGreen™ signal (IGH@) on the normal chromosome 14. The interphase cell shows the standard signal pattern of 2 fusions, 1 red and 1 green, that denotes the presence of the balanced t(11;14)(q13;q32) (Probe: LSI IGH/CCND1 dual-colour dual-fusion translocation probe, Vysis)
The most common entity from which MCL should be distinguished is CLL as they may have over-lapping features, both being CD5-positive small B-cell neoplasms. Reports in the older literature of t(11;14) in CLL almost certainly referred to cases of MCL. A case of splenic lymphoma with histology and flow cytometry results that could not distinguish between SMZL and a MCL was confirmed as MCL with the demonstration of IGH@/CCND1 fusion signals [133]. Ho et al. identified 28 cases with typical flow MCL phenotype, but FISH showed that 9/28 did not contain the t(11;14) and had cytogenetic abnormalities commonly found in CLL. No significant immunophenotypic or morphological differences between t(11;14)-positive and negative cases could be discerned [134].
The t(11;14) is usually an obvious rearrangement on conventional cytogenetics, although complex translocations masking the t(11;14) have been described, requiring FISH with IGH@ and CCND1 probes to identify the fusion signals when the t(11;14) in not visible in an abnormal clone. Despite the complex nature of these rearrangements, there are usually abnormalities involving the 11q13 and 14q32 breakpoints, providing a clue as to the underlying genetic rearrangement [135]. However, reports have illustrated the need for FISH despite the finding of an abnormal karyotype with no evidence of any apparent abnormalities of either chromosome 11 or 14. FISH has shown an IGH@/CCND1 fusion signal on a “normal” chromosome 14, apparently caused by an insertion of a sub-microscopic segment of 11q13 into 14q32 [136, 137]. The Spanish Cooperative Group for Hematological Cytogenetics identified 145 cases of MCL at diagnosis with an abnormal karyotype. All contained either a t(11;14), a variant CCND1 translocation with a three-way translocation involving 11, 14 and a third partner chromosome, a variant translocation involving the IGL@ gene on chromosome 22 (t(11;22)), or a crypic t(11;14). Nine patients showed the IGH@-CCND1 fusion by FISH only, despite exhibiting an abnormal karyotype [138]. A blastoid variant of MCL did not contain a t(11;14) but had a deletion of 11q and abnormalities of 13p and 21p. Additional M-FISH and locus-specific FISH tests identified chromosome 11 and chromosome 14 sequences and CCND1-IGH@ fusion signals on both 13p and 21p [139]. An atypical t(11;14) was identified in 18/103 MCL cases and included patients with duplications of the CCND1-IGH@ fusion, three-way translocations and insertions, emphasising the importance of FISH to confirm unusual translocations [140].
As with most other translocations involving the immunoglobulin heavy chain locus, variant translocations involving one or other of the light chain genes have been reported. Wlodarska et al. reported three cases of the IGK@ variant, t(2;11)(p11;q13), with the 11q13 breakpoint mapped to the 3′ region of CCND1, in contrast to the 5′ breakpoint of the t(11;14) [141]. They further postulated that this variant may represent a more indolent form of MCL.
FISH, M-FISH and aCGH have identified extra abnormalities in 80–92 % of cases [132]. Additional abnormalities with recurrent breakpoints are listed in Table 44.2. There appear to be differences in the patterns of extra abnormalities between those cases shown to have primarily lymph node-based disease and those with peripheral blood disease, with the lymph node-based cases appearing more cytogenetically complex and more likely to show deletions and chromosome loss. Loss of 17p and gain of 3q have been associated with disease progression and shown to be independent indicators of poor prognosis in multivariate analysis [135, 142, 143]. A deletion of 11q22-23 has been observed in approximately 20–40 % of MCL, as is seen also in CLL, with two loci of deletion defined, one at 11q22.2 encompassing the ATM gene and another more distally at 11q23.3 [144].
Table 44.1
Frequency of translocations in MALT lymphomas
Site of MALT lymphoma | Translocation | Frequency (%) |
|---|---|---|
Stomach | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 57/256 (22 %) 8/256 (3 %) 1/256 (0.4 %) 8/208 (4 %) |
Lung | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 22/62 (42 %) 5/62 (8 %) 4/62 (6 %) 0/18 (0 %) |
Ocular Adnexae | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 8/110 (7 %) 0/110 (0 %) 14/110 (13 %) 4/66 (6 %) |
Salivary Gland | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 2/101 (2 %) 1/101 (1 %) 5/101 (5 %) 0/60 (0 %) |
Thyroid | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 1/18 (5.5 %) 0/18 (0 %) 0/18 (0 %) 3/13 (23 %) |
Skin | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 4/108 (4 %) 0/108 (0 %) 7/108 (6.5 %) 2/20 (10 %) |
Liver | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) | 0/10 (0 %) 0/10 (0 %) 5/10 (50 %) |
Intestine | t(11;18)(q21;q21) t(1;14)(p11;q32) t(14;18)(q32;q21) t(3;14)(p14.1;q32) | 3/20 (15 %) 2/20 (20 %) 0/16 (0 %) 0/10 (0 %) |
Blastoid and pleomorphic variants appear more likely to contain higher numbers of additional abnormalities, a higher frequency of 1p and 17p deletions as well as 10p abnormalities, a higher proliferation index and a shorter survival [143]. A cluster of genomic alterations identified by allelic genotyping, 1p21, 3q27, 7q22–36, 6p24, 9p21, 9q31 and 16p12, were closely associated with cell proliferation (Ki-67 staining) [143].
The overall number of deletions has been shown to be a significant independent prognostic marker, with 3 or more indicating poor clinical outcome [132]. Deletions of 11q23 and 6q21 have been associated with extranodal disease, and deletions of 13q14 and 6q21 were independent predictors of poor outcome in multivariate analysis [145]. MCL studied using metaphase CGH has confirmed the common regions of gain and loss and found gain of Xq and loss of 17p to be associated with a significantly worse prognosis [146, 147]. Deletions of TP53 have been demonstrated in a high proportion of MCL cases in leukaemic phase [148].
It has been suggested that the ability to identify the abnormal clone by conventional cytogenetics provides some prognostic information. Katzenberger et al. associated complex karyotypes with higher proliferation indices and an inferior prognosis. By conventional cytogenetics, MCL with more than two structural aberrations was characterised by a significantly worse outcome [132]. Woroniecka et al. found that the presence of near-tetraploidy and two copies of the t(11;14) were correlated with the blastoid variant of MCL and a poor prognosis. In the same study, the t(11;14) seen as a sole abnormality was associated with an intermediate clinical outcome and a normal result by conventional cytogenetics with a better prognosis [149]. However, the number of cases in this study was small.
Over 80 % of MCL cases show classical morphology with less than 15 % blastoid or pleomorphic subtypes [132]. Most cases of blastoid MCL have a complex karyotype with 3 or more chromosomal abnormalities in addition to the t(11;14). Four blastoid MCL cases were characterised by extra CCND1 copies identified by FISH [150]. Different patterns of additional abnormalities have been distinguished according to the morphological features of the blastoid mantle cells, with abnormalities of 13, 18 and 8 commonly seen in “classic” blastoid MCL and abnormalities of 13, 17 and 3 most common in the “pleomorphic” blastoid MCLs. Tumours with prominent nucleoli in the malignant cells have commonly contained chromosome 17 abnormalities [151].
The association of the t(11;14) and one of the Burkitt translocations is a rare but reported observation. Hao et al. reported five cases of MCL with 8q24 abnormalities in addition to the t(11;14), three of which were shown by FISH to have a rearrangement of MYC and two an extra copy of MYC [152]. All had blastoid morphological features and 4/5 had leukaemic involvement. Vaishampayan et al. also described two cases of blastic transformation of MCL associated with a very short survival with t(8;14) in addition to t(11;14) in one and dup(8q) with an extra copy of MYC in the other [153]. Further cases of the association between blastoid MCL and a Burkitt translocation have been reported [154–156], and quantitative PCR showed MYC over-expression to be significantly associated with shorter survival in MCL [157]. Another two patients with blastoid variant of MCL and t(11;14) plus MYC rearrangements showed Burkitt-like morphology [158]. Thus, MYC over-expression appears to be implicated in blastic transformation of MCL.
There is considerable dispute as to whether “indolent” MCL is truly MCL or rather a variant of CLL. Orchard et al. divided 80 patients with t(11;14) + MCL into those with peripheral lymphadenopathy (“nodal group”) and those without (“non-nodal group”) [159]. Median survival was significantly longer in the non-nodal group and of 6 long-term survivors (>90 months), all were non-nodal and the 5 whose IGHV mutation status had been determined all had mutated IGHV genes. Nodit et al. reported a single case of proven MCL (with an 11;14 translocation) who had had a previous lymph node biopsy 12 years previously that was shown to be cyclin D1+ on immunostaining [160]. The patient had not required therapy for most of the intervening years and clearly showed an indolent course. Another unusual report described a patient with both B-CLL, predominantly seen in the peripheral blood with classic immunophenotype, and MCL, shown by FISH to have the t(11;14) predominantly in lymph nodes, occurring concurrently [161].
A number of cyclin D1-negative MCL cases have been reported. Most of these have been identified as involving either the cyclin D2 locus (CCND2) at 12p13 or the cyclin D3 locus (CCND3) at 6p21.2 [162, 163]. The cryptic t(12;14)(p13;q32) involving the IGH@ locus or the t(2;12)(p12;p13) involving the IGK@ locus result in over-expression of cyclin D2 [164, 165], but immunostaining for cyclin D2 appears non-specific as it has been observed in most B-cell lymphomas [166]. The t(6;14)(p21.1;q32.3) involving the cyclin D3 locus has also been observed in other histological subtypes of mature B-cell malignancies [167]. Despite gene expression analysis revealing upregulation of D2 and D3 mRNA in cyclin D1-negative cases and showing good correlation with protein expression, interphase FISH has identified translocations or amplification of CCND2 (12p13) or CCND3 loci (6p21.1) in some but not all cases [162, 165]. Of note was the incidence of central nervous system (CNS) lymphoma in five of eight cases in one series of cyclin D1-negative MCL [163].
Diffuse Large B-Cell Lymphoma, NOS
DLBCL, not otherwise specified (NOS), are a group of B-cell neoplasms characterised by proliferation of large cells with a diffuse growth pattern. Estimated to make up approximately 40 % of all cases of NHL, DLBCL is one of the commonest types of lymphoma. The WHO entity is defined by inclusion of large B-cell lymphomas that do not meet the criteria for categorisation as particular DLBCL subtypes or other discrete B-cell neoplasms, and this makes for considerable clinico-pathological heterogeneity within this disease grouping.
Conventional cytogenetics has a high success rate and abnormality rate in DLBCL. Karyotypes in DLBCL are usually complex; greater than 90 % of cases have two or more abnormalities. In a study of 363 consecutive cases by Cigudosa et al., 72 % of abnormal karyotypes were hyperdiploid, 19 % were pseudodipoid and 9 % were hypodiploid [168]. Both translocations and copy number changes are prevalent in DLBCL. The most common translocations in DLBCL involve the immunoglobulin loci at 14q32 (IGH@), 22q11 (IGL@) and 2p12 (IGK@). A 14q32 breakpoint has been observed in approximately 50 % of cases analysed by conventional G-banding [168, 169]. Frequent non-random cytogenetic abnormalities are translocations involving BCL6, BCL2 and MYC, which have been reported in approximately 20–45 %, 20–25 % and 5–16 % of cases respectively [168–173]. Non-random recurrent copy number changes identified by metaphase CGH include gain of Xq, 2p, 7q, 12p and 18q and loss of 6q and 17p [174]. Array CGH identified gain 1q, 2p, 3p, 3q, 6p, 7p, 7q, 9p, 12q and 18q and loss of 1p13.1–p36.33, 6q16–24 and 17p12–p13.2 [175].
Cell of origin gene expression profiling (GEP) sub-classifies DLBCL into germinal centre B-cell type (GC), and activated B-cell type (ABC) or non-GC type [176, 177]. GC type DLBCLs are thought to arise from germinal centre B cells, whereas ABC type DLBCLs probably arise from post-germinal centre B cells arrested during plasmacytic differentiation. Patients with GC type DLBCL have significantly better responses to chemotherapy than patients with ABC type DLBCL, lending weight to the idea that the two groups represent discrete biological entities [177]. However, thus far, translation of GEP cell of origin findings to routine clinical practice has proved challenging. Immunohistochemistry to differentiate GC and ABC types of DLBCL has shown variable correlation with gene-expression profiles, but does not precisely reproduce the segregation of patients obtained using gene-expression signatures [178]. Meanwhile the addition of rituximab to chemotherapy regimens appears to improve survival in both GC and ABC DLBCL types, and it remains unclear if the cell of origin classification retains its prognostic power in the rituximab era [179–182]. However, it has become apparent that cytogenetic abnormalities in patients with DLBCL also tend to cluster according to the GC and ABC/non-GC status of the tumours.
The t(14;18)(q32;q21) results in deregulated expression of BCL2 due to juxtaposition of the BCL2 gene to the immunoglobulin heavy chain enhancer. It is present in over 85 % of cases of follicular lymphoma and is also observed in approximately 20 % of DLBCL cases. Variant translocations involving the immunoglobulin kappa and lambda loci may also occur. DLBCL cases with BCL2 translocations are associated with primary nodal disease and the GC type [177]. That is, most patients with a translocation involving BCL2 are GC type, but not all GC type DLBCL have BCL2 translocations. It has been proposed that DLBCL cases with a BCL2 rearrangement may represent transformation of previously unrecognised follicular lymphoma. Frequent secondary abnormalities in t(14;18) karyotypes from DLBCL are trisomy 7 and trisomy 12 [183]. BCL2 translocation was thought to be indicative of poor prognosis in DLBCL treated with CHOP chemotherapy alone by some investigators [171, 184]. However with the advent of combination rituximab and multi-agent chemotherapy, this no longer appears to be the case [185, 186]. Copy number changes also cluster according to the cell of origin. Using aCGH, Tagawa et al. found gain of 1q, 2p, 7q and 12q to be associated with GC type DLBCL [187]. Gain or amplification of REL or 2p has been consistently observed by a number of investigators [180, 188–190], as has gain of 12q [175, 180, 190, 191]. In a comprehensive study combining oligonucleotide aCGH and gene expression profiling in DLBCL, Lenz et al. [180] demonstrated differential use of oncogenic pathways by the GC and ABC types. In all, 12 % of patients with GC-type and no patients with ABC-type DLBCL had amplification of the MIR17HG microRNA cluster on chromosome 13. Although this cluster had been linked to DLBCL by other investigators, the study by Lenz et al. was the first to show that it is exclusively confined to the GC type. Other novel abnormalities associated with GC-type DLBCL were loss of PTEN on chromosome 10 (homozygous loss of PTEN was not identified in any ABC-type tumours) and deletion of the TP73 gene on chromosome 1p. Interestingly homozygous PTEN deletion was associated with the presence of the t(14;18).
Meanwhile, translocations involving BCL6 have been linked to cases of extranodal lymphoma and the ABC (or non-GC)-type DLBCL [176, 177]. The most common translocation involving BCL6 at 3q27 is the t(3;14)(q27;q32) driving BCL6 expression as a result of juxtaposition of the gene to the region of the immunoglobulin heavy chain enhancer. Reciprocal translocations between it and the light chain loci and other non-immunoglobulin gene partners have also been reported [192–194]. In DLBCL cases where the t(3;14) is identified by conventional karyotyping, recurrent secondary abnormalities are gain of chromosomes X, 11 and 13, and deletion of 6q and 8p23 [183]. The impact of BCL6 translocation on outcome in DLBCL is unclear. Most studies predate the rituximab era and variously report favourable [170, 195, 196], neutral [197] or adverse [171] prognostic significance. Since CHOP-R has replaced CHOP as first-line therapy for DLBCL, it appears that BCL6 rearrangement has lost any independent prognostic impact [186, 198]. Copy number changes associated with the ABC/non-GC cell of origin by several investigators include trisomy 3/gain of 3q, gain/amplification of 18q and 19q and loss of 6q and 9p21 [177, 180, 184, 187, 191, 199, 200]. 18q amplification results in frequent over-expression of BCL2 mRNA and protein in the APC/non-GC type and was reported to be a poor prognostic variable in some studies [174, 201] but not in others [175, 180, 190]. Another random non-recurrent genetic abnormality in the ABC type is mutation of CARD11 resulting in activation of NFKB signalling, and Lenz et al. reported that CARD11 mutations and trisomy 3 tended to co-segregate in the small number of patients who had a CARD11 mutation included in their dataset [180]. In this study, ABC DLBCL patients with trisomy 3 or a 9p21 deletion (involving CDKN2A locus), or both had significantly worse outcomes than ABC-type patients without these abnormalities. Other investigators have also reported that patients with 9p21 deletion had adverse outcomes in the CHOP [187] and R-CHOP era [202].
One important prognostic cytogenetic variable in DLBCL is MYC translocation. While MYC translocation are the hallmark of Burkitt lymphoma (BL), they are not unique to BL and can be observed in other B-cell lymphomas including CLL, DLBCL, B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma (inter-DLBCL/BL), and plasmablastic lymphoma. The WHO classification restricts BL to tumours with a MYC translocation involving immunoglobulin loci (IGH@ at 14q32, IGK@ at 2p12 or IGL@ at 22q11) in a simple karyotype. Simple karyotype is defined as no or only a few additional changes by conventional cytogenetics or less than 6 abnormalities detected by aCGH based on the seminal findings of Hummel et al. [203]. To date, the guidelines have proved somewhat challenging to interpret in clinical practice as aCGH technology is yet to be widely taken up outside the research or clinical trial setting and the definition of “only a few” is somewhat imprecise. A recent study suggests that cytogenetically, distinguishing cases of BL from inter-DLBCL/BL (mean additional abnormalities 1.7 and 3.3 respectively) is likely to be more difficult that distinguishing these two entities from DLBCL cases which displayed far more genetic complexity (mean additional abnormalities 21.7) [204]. From these studies, it is clear that measures of karyotypic complexity play a role in the diagnostic decision-making algorithm and complement the information obtained from FISH testing in aggressive B-cell lymphomas. Thus an effort should be made to incorporate conventional metaphase karyotyping or molecular karyotyping by aCGH into the work up of lymphomas where there is a high pretest probably of a MYC translocation.
A recent study measuring the frequency of MYC, BCL6 and BCL2 rearrangements in a large population of patients with de novo DLBCL using i-FISH found that the incidence of MYC rearrangement was 14 % [186]. MYC rearrangement was rarely present as the sole translocation (only 2 % of cases); almost all tumours also had a coexisting 14;18 translocation, a BCL6 rearrangement of both [186]. MYC rearrangement per se is associated with a poor prognosis in DLBCL in most published series [200, 205–208]. The so-called “double-hit” and “triple-hit” lymphomas, high-grade B-cell lymphomas with a BCL6 rearrangement and/or a BCL2 rearrangement in addition to a MYC translocation that fall into the DLBCL, NOS category on the basis of their morphology and immunophenotype, are also generally acknowledged to have a poor prognosis [203]. At this stage, it is unclear if the MYC translocation partner has prognostic significance. Johnson et al. found that involvement of non-immunoglobulin partners (non-IG-MYC) was common in DLBCL and these patients tended to have a more favourable outlook than patients with IG-MYC translocations [209]. However, in the study by Barrans et al. involving patients treated with R-CHOP, any MYC rearrangement as a single parameter remained an independent indicator of poor prognosis [186]. In the study by Seegmiller et al. that excluded non-IG-MYC translocations, only 1/4 “double-hit” lymphomas (DHL) was classified as a DLBCL; the other 3 were inter-DLBCL/BLs [204]. Despite the caveat that there is some inherent biological heterogeneity in the variably defined “double-hit” DLBCL group, on the whole it does appear that these patients are less likely to respond satisfactorily to R-CHOP regimens [186, 205, 209, 210]. Yet studies of “double-hit” DLBCL have failed to demonstrate a survival benefit for dose-intensified chemotherapy regimens such as those favoured for the treatment of BL [193, 205, 211, 212]. Hence such patients are currently candidates for investigational approaches under the aegis of clinical trials.
T-Cell/Histiocyte-Rich Large B-Cell Lymphoma
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a morphological variant of DLBCL characterised by a small number of large CD20-positive B cells surrounded by an abundant T-cell infiltrate and usually accompanying histiocytes. Morphologically these cases bear some resemblance to cases of nodular lymphocyte predominant Hodgkin lymphoma (NLPHL). Due to the relative paucity of neoplastic cells, successful identification of the malignant clone by conventional cytogenetics is problematic. Only two cases with abnormal karyotypes have been reported. Both karyotypes had a near-tetraploid modal number and were complex with numerical and structural abnormalities but no specific abnormality [213, 214]. MYC, BCL2 or BCL6 rearrangements were not detected. Interestingly, the case reported by De Laval et al. [213] had an isochromosome of 1q, a cytogenetic abnormality that is also frequently observed in NLPHL [215]. Franke et al. performed CGH on DOP-PCR amplified DNA isolated from micro-dissected CD20-positive cells taken from 17 THRLBCL tumours [216]. Copy number changes were present in all cases. The most frequent changes were gain of Xq, 4q, Xp and 18q21 and loss of 17p. On average, THRBCL had less copy number changes than NLPHL cases (4.7 vs. 10.8). However, the authors had noted similar patterns of copy number change in NLPHL tumours previously [217], including gain of 4q that is an unusual finding in other types of lymphoma, leading to speculation that THRLBCL and NLPHL may share the same cell of origin.
Primary DLBCL of the CNS
Primary DLBCL of the CNS (CNS DLBCL) is an aggressive B-cell lymphoma restricted to the CNS. Investigation of CNS DLBCL by i-FISH has revealed that chromosomal translocations are common in this disorder. They frequently involve the immunoglobulin genes [218, 219]. In a FISH study by Montesinos-Rongen et al., rearrangements of IGH@, IGK@ and IGL@ were present in 23 %, 24 % and 36 % of cases respectively [219]. BCL6 translocations are also frequent, occurring in up to 47 % of cases and do not necessarily involve immunoglobulin partners [218–220]. Non-immunoglobulin translocation partners for BCL6 identified to date include HIST1H4I, HSPCA and GAPDH [220, 221]. The t(8;14), t(11;14) and t(14;18) have not been identified in CNS DLBCL [219], but MYC rearrangements by FISH were present in 3 % of cases studied by Cady et al. [218]. Gain or amplification of 18q21 is frequent [219, 222]. In addition to 18q21 gain, Weber et al. also identified recurrent loss of 6q and gain of 12q, 22q by CGH [222].
In 2009 Schwindt et al. [223] analysed 19 tumours using a high-density SNP array. Regions of recurrent loss were identified at 3p14.2, 4q35.2, 6p21.32, 6q21, 8q12–12.2, 9p21.3, 10q23.2 and 12p13.2 and there were recurrent copy number gains of 18q21–23, 19q13.31 and 19q13.43 together with gain of the X chromosome and trisomy 12. There was also recurrent copy number neutral loss of heterozygosity (CN-LOH) at 6p and 9p21.3. Deletion or CN-LOH involving 6p21.32, the site of the HLA Class II genes, was found in 73 % of tumours and was the most frequent abnormality detected. Earlier work had also identified deletions at this site as common abnormalities in CNS DLBCL [224–227]. A functional consequence proposed to result from loss of HLA class II expression through homozygous deletion or CN-LOH is tumour escape from immune surveillance and absent HLA class II correlates with unfavourable clinical outcomes in some studies [228, 229]. Del(6)(q21), the next most frequent abnormality, was present in 53 % of cases and is common to DLBCL NOS and other B-cell lymphomas. These findings validate the work of other investigators who have previously reported recurrent loss of 6q, gain of 18q and copy number loss at 9p21.3 in CNS DLBCL using alternative molecular cytogenetic techniques [218, 219, 222, 225, 230, 231].
Gain of 18q21 results in additional copies of the BCL2 and MALT1 genes and both have been implicated in disease pathogenesis. However, more recent studies indicating that NFKB pathway activation is ubiquitous in this condition [232] suggest that perturbation of NFKB activity through MALT1 amplification may be a critical oncogenic event in CNS DLBCL. Similarly, frequent deletion or silencing of CDKN2A at 9p21 indicates that it is likely to be a critical TSG that must be bypassed for CNS DLBCL lymphomagenesis [230, 233, 234]. In the study of 75 immunocompetent patients with CNS DLBCL by Cady et al., del(6q) was an independent predictor of poor prognosis [218].
Primary Cutaneous DLBCL, Leg Type
Primary cutaneous DLBCL, leg type (PC LBCL) is a large B-cell lymphoma of the skin that often presents on the leg in elderly women with frequent secondary dissemination. FISH studies in small numbers of patients have indicated that IGH@ translocations are common in this disorder (6/11 patients) [235]. MYC and BCL6 rearrangements have been reported in 6/14 and 2/14 cases respectively [236]. Copy number changes consistently identified by CGH include gain of 18q and loss of 6q [237].
Dijkman et al. compared 12 cases of PC LBCL to 19 cases of primary cutaneous follicle centre lymphoma (PCFCL) using BAC aCGH and noted distinct patterns of genetic imbalance in the two disorders. Other investigators also noted differences in copy number changes in PCFCL and PC LBCL by CGH [237], supporting their classification as discrete biological entities. In the study by Dijkman and colleagues [238], amplification of 18q was present in 67 % of PC LBCL cases and involvement of the BCL2 and MALT1 genes was confirmed by i-FISH. Loss of 9p21.3 was identified in 41 % of PC LBCL cases and was unique to the PC LBCL subset. Although the number of cases was small, detection of del(9)(p21) by BAC aCGH or CDKN2A gene silencing by DNA methylation was associated with poor prognosis disease in this study. A follow-up study by Senff et al. [239] mapped 9p21 deletions by multiplex ligation-dependent probe amplification (MLPA) and measured CDKN2A methylation in a larger series of 64 cases and found deletion to be more common than methylation. Five-year disease-specific survival was 43 % for cases with a 9p21/CDKN2A abnormality as opposed to 70 % for those without (p = 0.06).
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBL) is a B-cell lymphoma presumed to be of thymic B-cell origin arising in the mediastinum without nodal or bone marrow involvement. Compared to DLBCL, it is more frequent in young women and has a more favourable prognosis. PMBL bears a strong resemblance to classical Hodgkin lymphoma (CHL) clinico-pathologically, and the so-called grey-zone lymphoma with features intermediate between the two conditions can pose a diagnostic dilemma.
Early cytogenetic studies identified gain of 9p, 12q and Xq as recurrent cytogenetic abnormalities in PMBL [240–242]. Bentz et al. [240] investigated 43 cases by CGH and observed gains of 9q and Xq in 40 % or more. Gain of Xq, 9p21 and 12q14 was confirmed in 87 %, 75 % and 35 % of cases respectively using more sensitive i-FISH testing. For other types of B-cell lymphoma assayed by FISH, the rate of 9p gain was very low (6/308 cases), identifying 9p gain as a distinctive feature of PMBL. Palanisamy et al. published the results of conventional karyotyping in 17 patients with PMBL. Clonal cytogenetic abnormalities were present in 11/17 (65 %) cases. The karyotypes were hyperdiploid or near-tetraploid in the majority of cases with complex numerical and structural abnormalities and no specific aberration. A t(14;18)(q32;q21) was present in two cases and one case had a t(3;14)(q27;q32). CGH results for 40 PMBL cases were also compared with 91 cases of DLBCL. Gain of 9, 19 and X and loss of chromosome 4 were significantly more frequent in the PMBL cohort [243]. Bea et al. compared the chromosome CGH profiles of tumours that had been classified into the ABC and GC subtypes of DLBCL and PMBL by gene expression profiling and confirmed 9p gain and 2p14–16 gain as distinctive findings in PMBL [191].
BAC aCGH studies in PMBL have been performed by Weesendorf et al. [244] and Kimm et al. [245]. Wessendorf et al. studied 37 cases and reported recurrent gain of 9p24 (68 %), 2p15 (51 %), 7q22 (32 %), 9q34 (32 %) and 12q (30 %) [244]. Candidate oncogenes REL and BCL11A were included in a region of high-level amplification on 2p in this study; however, the REL locus was not part of a larger minimal consensus region of 2p gain. Kimm et al. [245] performed BAC aCGH on samples from 20 patients with PMBL. Fifty-one regions of recurrent genomic imbalance were detected involving 1p, chromosome 2, chromosome 3, 4q, chromosome 5, 6q, 7p, chromosome 8, 9p, 11q, chromosome 12, 17p, chromosome 20, 21q and the X chromosome. Novel regions of gain were 2q13, 3q23, 3q26.1–q27.3, 5q33.3–q34, 7p12.3–p14.1 and 7p22.1–p22.3. Consistent with prior studies, the REL locus was frequently gained or amplified and there were frequent gains on the X chromosome. JAK2 copy number was increased in 62 % of cases, although the region of highest recurrence mapped to 9p13.1–p13.3 and excluded the JAK2 locus. Recurrent regions of loss were 1p13.1–p13.2, 3p, 4q, 6q and 17p [245]. Weniger and colleagues [246] proceeded to investigate the consistently identified 2p14–p16 copy number gain in greater detail. A FISH probe hybridising to the BCL11A locus identified gain or amplification in 75 % of tumours tested. The number of copies of the BCL11A gene also correlated with transcript abundance. Meanwhile REL gain amplification was present by FISH in a similar proportion of cases, and despite the fact that transcript levels were not consistently increased in cases with increased gene dosage, REL copy number increases were associated with nuclear accumulation of the REL protein [247].
In 2008 Lenz et al. [180] used combined oligonucleotide aCGH and gene expression profiling data to compare ABC and GC subtypes of DLBCL with PMBL. The authors identified 30 minimal common regions (MCRs) with subtype distinction. MCRs that occurred with the highest frequency in PMBL were del(6q), 9p amplification or gain, monosomy 10 and 20p gain or amplification. The best discriminator for PMBL was 9p amplification. It was observed in 45 % of PMBL, but only 11 % and 7 % of ABC and GC DLBCL subtypes respectively. Genes encoding JAK2 and PDL2 were the most strongly upregulated in the 9p MCR [180, 248]. Meier et al. [249] reported 9p24 gain in 33 % of PMBL cases studied by FISH in a tissue microarray study of lymphoma. Amplification of 9p is also common in CHL and was investigated further using cell lines and primary tumours from patients with CHL and PMBL. Underscoring the importance of oncogenic JAK/STAT signalling in the pathogenesis of these two conditions is the finding that SOCS1 loss of function mutations resulting in sustained phosphorylation of JAK2 and deregulated STAT activity are present in approximately 50 % of PMBL and CHL tumours [250, 251]. With respect to PDL1/PDL2 amplification in PMBL, Green et al. [252] showed that PDL1 at 9p24.1 was amplified in 63 % (26/41) cases of PMBL and that amplification correlated with transcript abundance. There was also over-expression of PDL2 in tumours from PMBL patients with 9p24.1 amplification using immunohistochemistry. In conclusion, taking into consideration the fact that PDL1 and PDL2 have been implicated in tumorigenesis through reduced immune surveillance, they are also attractive candidate oncogenes within the region of 9p characteristically amplified in the related disorders of PMBL and CHL.
ALK-Positive Large B-Cell Lymphoma
The ALK-positive large B-cell lymphomas represent a rare entity that frequently exhibits plasmablastic morphology. Translocations involving the ALK gene on 2p23 occur generally in T-cell or null-cell phenotype lymphomas (discussed below in the Mature T- and NK-cell Neoplasms section). In 1997, Delsol et al. reported seven patients with large B-cell lymphoma that showed cytoplasmic ALK staining but no evidence of the t(2;5)(p23;q35) that is observed in the majority of ALK-positive T-cell or null-cell anaplastic large cell lymphomas [253]. Western blotting of one case showed that the full length ALK was expressed. Subsequent reports have identified a handful of cases of B-cell lymphomas carrying the t(2;5) and expressing the NPM-ALK fusion [254–256]. However, the majority of ALK-positive B-cell lymphomas in both adults and children contain a variant translocation, the t(2;17)(p23;q23), involving the Clathrin gene (CLTC) at 17q23, which is associated with a distinctive fine granular cytoplasmic localisation of the ALK staining on immunohistochemistry [257–262].
Cryptic ALK rearrangements have also been observed, with insertions of 3’ALK into the NPM locus, creating a sub-microscopic NPM-ALK fusion and insertion of 5’SEC1A, a gene located at 4q21 that has not been previously reported in ALK fusions in lymphoma, upstream of the 3′ end of ALK [ 255]. The SEC31A-ALK fusion was also associated with granular cytoplasmic ALK staining.
Burkitt Lymphoma
Burkitt lymphoma (BL) is an aggressive mature B-cell lymphoma of children and adults with a characteristic clinical presentation, morphology and immunophenotype. Juxtaposition of MYC to an immunoglobulin enhancer (IG-MYC translocation) resulting in deregulated expression of the MYC proto-oncogene is the hallmark of this condition. In 85 % of cases, the immunoglobulin heavy chain (IGH@) at 14q32 is the partner gene, resulting in the reciprocal t(8;14)(q24;q32) (Fig. 44.1). In the remaining 15 % of cases, the translocation involves IGK@ at 2p11 or IGL@ at 22q11, resulting in the t(2;8)(p11;q24) or the t(8;22)(q24;q11) variant [263]. Although MYC translocations are strongly associated with BL, they are not specific for this condition and are encountered in other aggressive B-cell lymphomas, albeit at lower frequency. The distinction is that in BL, there is strong evidence to support IG-MYC translocation as the primary or initiating event in lymphomagenesis, whereas in other B-cell disorders MYC rearrangement is more likely to occur during evolution of the neoplastic clone. Furthermore, MYC translocation is not absolutely required in BL because a small percentage of cases will meet stringent morphological and molecular criteria for BL in the absence of a demonstrable MYC translocation. In some cases, this may be explained by methodological limitations. For example screening for MYC rearrangement using a FISH break-apart probe (Fig. 44.3) may miss both unusual translocations where the breakpoint on 8q is a long way upstream or downstream of the MYC locus, and small insertion events. In other cases, emerging evidence suggests that alternative genetic events, such as those leading to downregulation of the microRNA MIR34B, can mimic the molecular and immunophenotypic consequences of IG-MYC translocation [264].
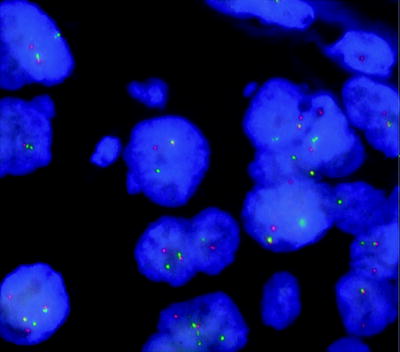
Fig. 44.3
Paraffin-embedded tissue section from the lymph node of a patient with Burkitt lymphoma. A MYC break-apart probe (LSI MYC dual-colour break-apart rearrangement probe, Vysis) shows MYC signal rearrangements in most cells, indicating the presence of a translocation disrupting the MYC locus at 8q24. The fusion signal indicates an intact MYC locus, and the red and green signals indicate the presence of a translocation splitting the SpectrumOrange™ labelled 5′MYC and SpectrumGreen™ labelled 3’MYC signals apart. This result does not identify the partner gene involved in the translocation with MYC
A gene expression profiling study by Dave et al. demonstrated that cases of BL with typical and atypical morphology were interchangeable at a molecular level. Higher expression of MYC target genes and germinal centre B-cell genes distinguished cases with a t(8;14) from other lymphomas and, together with reduced expression of genes in the NFKB pathway and MHC Class I genes, reliably distinguished BL from DLBCL with or without the t(8;14) [210]. A contemporaneous study by Hummel et al. [203] involved comprehensive characterisation of mature aggressive B-cell lymphomas using gene expression profiling, BAC/PAC aCGH and i-FISH for MYC, BCL2 and BCL6 translocations. Consistent with the findings of Dave et al. [210], there was also striking homogeneity of BL cases at a molecular level including cases with atypical morphology.
Using a set of 58 differentially expressed genes, Hummel et al. [203] proceeded to define a molecular gene signature of BL and used it to classify lymphomas into three groups: molecular BL (mBL), non-molecular BL (non-BL) and intermediate. On the basis of i-FISH and aCGH findings, the same cohorts were also assigned a molecular karyotype. “MYC-simple” was defined as the presence of a MYC rearrangement resulting in juxtaposition to an immunoglobulin enhancer, less than six copy number imbalances by aCGH (low genomic complexity score) and the absence of an IGH@-BCL2 fusion or a BCL6 rearrangement. “MYC-complex” comprised either cases with a non-IG-MYC translocation, or an IG-MYC translocation together with a high genomic complexity score (six or more genomic imbalances), or a t(14;18) translocation or a BCL6 rearrangement or any combination of the three. The “MYC-negative” group comprised all those remaining patients without evidence of a chromosomal rearrangement involving MYC at 8q24 by FISH. Eighty-eight percent of patients with mBL had an IG-MYC translocation and 91 % had evidence of a MYC rearrangement, indicating that that absence of a MYC translocation need not preclude the diagnosis of BL if the clinical presentation, morphology and immunophenotype are otherwise classical.
There was a strong correlation between the mBL gene expression signature and the “MYC-simple” karyotype. Those patients with an intermediate gene expression profile mostly had “MYC-complex” karyotypes, but occasional cases were “MYC-simple” or “MYC-negative.” The majority of cases with a non-mBL gene expression signature had “MYC-negative” karyotypes. Cases with “MYC-simple” karyotypes had significantly better outcomes than the combined “MYC-complex” and “MYC-negative group” (5-year OS 70 % vs. 40 %, p = 0.005). In contrast, in patients with an intermediate or non-mBL gene signature, the presence of a MYC rearrangement was associated with a poor prognosis [203].
In order to place the findings of Hummel et al. [203] in the context of conventional metaphase cytogenetic studies, Boerma et al. [263] systematically analysed cases listed in the Mitelman Database of Chromosome Aberrations in Cancer. They defined “true” BL using morphological criteria, presence of an IG-MYC translocation and absence of a BCL2, BCL6 or CCND1 translocation. Variant IG-MYC translocations were slightly more common in adults than children, and adults were more likely to have gain of 8q, but in many other respects the karyotypes were very similar, supporting the concept that BL constitutes the same biological entity in children and adults. “True” BL was again characterised by low karyotypic complexity. In 40 % of cases, an IG-MYC translocation was the sole abnormality detected. Of the 205 paediatric cases studied, unbalanced chromosomal abnormalities were present in 60 %: one imbalance was present in 33 % of cases, two imbalances in 16 % and more than two in the remainder. Seegmiller et al. [204] also studied the karyotypic complexity of 34 patients with IG-MYC translocations classified according to WHO 2008 criteria. The average number of cytogenetic imbalances was 1.7 in patients with BL, 3.3 in inter-DLBCL/BL patients, 6.7 in patients with plasmablastic lymphoma and 21.7 in DLBCL. Patients with a simple karyotype, defined as less than or equal to 2 additional cytogenetic abnormalities, had a significantly greater likelihood of having a BL diagnosis. Therefore the investigators proposed that a cytogenetic imbalance score, using a cut-off value of 2, may be a valuable discriminator between BL and inter-DLBCL/BL cases.
Additional secondary abnormalities noted by Boerma et al. [263] in BL karyotypes were gain of 1q, predominantly as the result of a duplication event, trisomy 7 and trisomy 12; losses of 6q, 17p and 13q32-34 were less frequent. Gain of 1q was most often noted as a single additional cytogenetic abnormality, but apart from this no clear patterns of secondary abnormality were identified. The pattern of secondary abnormalities in metaphase preparations is in keeping with patterns of genomic imbalance observed by CGH, aCGH and FISH techniques in BL [265]. No consistent prognostic significance has been attached to secondary abnormalities. Gain of 1q and 7q and loss of 13q have variously been reported to be associated with inferior outcomes by some investigators [266–268], but there is a dearth of large studies involving uniformly treated patients able to address this question. Nelson et al. [269] studied 90 patients in a multi-centre Children’s Cancer Group study to assess the prognostic importance of del(13q) and gain of chromosome 7 in BL in children. FISH testing using a MYC break-apart probe, a 13q probe and a chromosome 7 centromere probe were performed on paraffin-embedded tissue (Fig. 44.3). MYC was rearranged in 93 % of patients in keeping with results in the earlier study by Hummel et al. [203]. Del(13q) and trisomy 7 were identified in 42 % and 10 % of cases respectively. Interestingly the frequency of 13q deletions in this study was far higher by FISH than by conventional cytogenetics, suggesting the existence of additional sub-microscopic deletions or the presence of non-proliferating sub-clones not detected by examination of metaphase preparations. Although numbers were small, the presence or absence of a MYC translocation or an additional signal for the chromosome 7 centromere made no difference to outcome; however, del(13q) detection by FISH was associated with significantly reduced 5-year overall survival [269].
B-Cell Lymphoma, Unclassifiable, with Features Intermediate Between DLBCL and Burkitt Lymphoma
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma (inter-DLBCL/BL) includes cases of high-grade B-cell lymphoma that do not meet the criteria for Burkitt lymphoma or DLBCL because of intermediate morphology, discordance between morphology and immunophenotype or for other clinical and biological reasons. Patients who meet the criteria for DLBCL who have a MYC translocation and patients who meet the criteria for Burkitt lymphoma in the absence of a MYC translocation are excluded from this category.
Between 35 and 50 % of inter-DLBCL/BL have a demonstrable MYC rearrangement but in contrast to Burkitt lymphoma (BL), many of these involve non-immunoglobulin translocation partner genes or are variant IG-MYC translocations involving IGK@ or IGL@ [203, 209]. It may be that in these cases, the MYC translocation occurs as a secondary event at the time of disease progression, with an IGH@ translocation less likely due to a preceding primary translocation between IGH and another oncogene such as BCL2 at the time of disease initiation [270, 271]. Seegmiller et al. [204] reported that karyotypes from patients with inter-DLBCL/BL had a mean complexity score of 3.3, whereas BL cases had a score of 1.6, suggesting a cut-off value of 2 as a useful discriminator in “grey-zone” cases with an IG-MYC translocation. Fifteen percent of all inter-DLBCL/BL cases have a BCL2 translocation, and inter-DLBCL/BL is enriched for patients with “double-” or “triple-hit” lymphomas which harbour translocations involving MYC and BCL2 and/or BCL6 simultaneously [203, 204, 263, 272]. The incidence of DHL in a recent large study of 1,260 cases of high-grade lymphoma was 4 %; [209] approximately two-thirds fitted the criteria for inter-DLBCL/BL and most of the remainder were DLBCL. In that subset of DHL cases harbouring an IG-MYC translocation rather than a non-IG-MYC translocation, studies have consistently found that “double-hit” patients were more likely to be older than patients with BL, have a variant IG-MYC translocation and have greater genetic complexity [203, 204, 263, 272]. Additional non-random, recurrent abnormalities identified by conventional cytogenetics in DHL cases were trisomy 7, 8, 11, 12, 18, 20, an additional X chromosome and loss of 3q27–29, 6q and 15q26 [263].
In the DHL cohort of inter-DLBCL/BL and DLBCL cases studied by Johnson et al., the presence of a non-IG-MYC translocation was associated with a slightly more favourable outcome, although morphology and IPI remained more powerful predictors of outcome [209]. In other studies, unstratified DHLs whether classified as inter-DLBCL/BL or DLBCL are reported to have a poor prognosis that is not modified by DLBCL regimens incorporating rituximab or dose-intensified BL treatment protocols [211, 212, 270, 272, 273]. Hence these patients are candidates for clinical trials designed to explore optimal therapeutic strategies.
Apart from MYC and BCL2 translocations in inter-DLBCL/BL, Pienkowska-Gela et al. [274] described four cases with a dup(11)(q23q13) and no evidence of MYC translocation. It is possible that duplication of material on 11q represents a novel, recurrent, non-random abnormality in patients in this diagnostic category, but this is yet to be verified in large series.
Mature T- and NK-Cell Neoplasms
T-Cell Prolymphocytic Leukaemia
T-cell prolymphocytic leukaemia (T-PLL) is a rare neoplastic disorder of post-thymic T cells. Clinically the presentation is generally with circulating leukaemia and massive splenomegaly. Often the WCC is greater than 100 × 109/L, which means there is generally little difficulty in obtaining tumour-enriched material from blood or bone marrow suitable to culture for karyotyping.
Cytogenetically, the disease is characterised by the inv(14)(q11.2q32) (Fig. 44.1) or the t(14;14)(q11.2;q32) which is present in approximately 80 % of cases [275–279]. These rearrangements place genes within the TCL1 locus under transcriptional control of the enhancer region of the T-cell receptor alpha delta (TRA@/D@) locus resulting in deregulated TCL1 expression. Where 14q32 rearrangements are absent, the t(X;14)(q28;q11.2), which translocates MTCP1 (a TCL1 homologue) to TRA@/D@ with similar biological consequences, is usually present [275, 277, 278, 280]. Interestingly, patients with the inherited disorder ataxia telangectasia (AT) are predisposed to T-cell malignancy, and the inv(14)/t(14;14) or the t(X;14) can be detected in small premalignant clones from these patients, suggesting that TCL1/MTCP1 deregulation is an early or initiating event during T-cell transformation but is insufficient for development of fully established malignancy [281]. In keeping with the concept that additional genetic changes are probably required for neoplastic progression, sporadic T-PLL karyotypes are usually complex at diagnosis and typically share a distinctive pattern of secondary cytogenetic abnormalities, including abnormalities of chromosomes 6, 8, 11, 12, 17 and 22 [275, 277, 279].
Changes in chromosome 8 are the most common secondary abnormalities and usually result in loss of material on 8p and trisomy for 8q, often as the result of formation of an isochromosome of 8q, an isodicentric chromosome 8p11, or a der(8)t(8;8)(p12;q11.2) [275, 278, 279, 282]. Monosomy 11 or deletion of 11q is the next most frequent abnormality and encompasses the ATM gene, which is generally considered the candidate TSG targeted by this deletion in T-PLL [277, 283]. Unsurprising, given the association between T-PLL and AT, mutations in ATM are also frequent in sporadic cases, such that there is usually bi-allelic ATM loss of function in this condition even in those patients without 11q22–q23 deletion [283–286]. Deletion of 6q, 12p, monosomy 13/del(13q), monosomy 17/del(17p) and monosomy 22/del(22q) are also seen by conventional karyotyping and/or FISH studies at lower frequencies [275, 277, 287–292]. CGH analysis has identified additional gains on 5p, 6p, 14qter, 21 and 22q12-qter, additional losses on 7q, 9p, 11p, 16q, 17q, and enabled the CDR involved in the del(11q) to be mapped in finer detail [279, 290, 293]. 50 K SNP array analysis of five patients with inv(14)/t(14;14) T-PLL confirmed recurrent gains in 6p and 8q and losses in 6q, 8p and 11q. Furthermore, large novel recurrent areas of loss in 10p and 18p were identified and large regions of uniparental disomy (UPD) on 3q (that were overlapping in two patients), 6p, 9p, 11q and 13q were also reported [290].
T-Cell Large Granular Lymphocytic Leukaemia
This is a T-cell disorder with a distinct clinical presentation, cell morphology and commonly a CD3+ CD8+ CD57+ TCRαβ+ immunophenotype. T-cell receptor gene rearrangement studies are generally used to demonstrate clonality and in most patients, the disease follows an indolent clinical course. A large recent Japanese case series reported normal karyotypes in 33/35 patients; one elderly male patient had a—Y clone and one patient had a structural abnormality of 14q [294]. Similarly, Dhodapkar et al. reported a low rate of cytogenetic abnormalities in their series [295]. There are less than 30 published abnormal karyotypes from patients with this condition in the literature and no consistent pattern of chromosomal derangement is apparent.
Reports are almost exclusively confined to conventional cytogenetic analysis; studies applying molecular techniques including FISH, metaphase CGH, aCGH and SNP-A are lacking. Most karyotypes have a near-diploid modal number and range in complexity from simple numerical aberrations and balanced reciprocal translocations to cases with multiple numerical and structural abnormalities [296, 297]. Man et al. have suggested that monosomy of chromosome 6 or del(6q) may be over-represented in this patient group [296], although deletion of 6q is not unique to this entity.
Chromosomal breakpoints involving the TRG@ and TRA@/D@ loci at 7p14–p15 and 14q11.2 respectively are described in patients with this disorder, including some cases of inv(14) or an X;14 translocation involving the TRA@/D@ locus that are more commonly associated with a diagnosis of T-PLL [297]. Distinguished by a CD3+ CD8+ CD56+ CD57- immunophenotype, a poor prognosis variant of T-cell LGL with a tendency to behave more like aggressive NK cell leukaemia has also been proposed to exist by some investigators [298]. The presence of an isochromosome 7 has been reported in 3/13 cases of aggressive T-cell LGL described to date [298–302].
Chronic Lymphoproliferative Disorder of NK Cells
Chronic lymphoproliferative disorder of NK cells (CLPD-NK) is an indolent disorder in which there is an expanded population of CD3-, CD16+ NK cells for greater than 6 months in the absence of an identifiable cause. In retrospective case series of patients subsequently shown to have an indolent clinical course, a normal karyotype at diagnosis is usual [294, 303–305]. T-cell receptors are by definition in the germ line configuration, meaning historically there has been a reliance on X-chromosome inactivation to demonstrate clonality in this disorder [305]. Although the presence of a cytogenetic abnormality does not automatically preclude a diagnosis of CLPD-NK, transformation to aggressive LGL leukaemia after 20 months was described in a patient with a mature NK cell lymphocytosis in whom a trisomy 8 clone was present at diagnosis. Evolution of his trisomy 8 clone was co-incident with blastic transformation [306].
Aggressive NK Cell Leukaemia
This rare CD2+, surface CD3- (sCD3-), CD56+ NK cell neoplasm with cytoplasmic CD3ε expression is primarily geographically restricted to Asia, Central and South America, strongly associated with EBV infection and usually involves the peripheral blood, bone marrow, liver and spleen. Although it has a distinct anatomical location and there are differences in patient demography, aggressive NK cell leukaemia (ANKL) appears to share some biological features with extranodal NK/T-cell lymphoma, nasal type (ENK/TL-NT). Hence, given their relative scarcity, patients with ENK/TL-NT and ANKL tend to be combined in case series where ANKL cases are usually in the minority. Biopsy material is frequently necrotic in this disorder, rendering it unsuitable for cytogenetic assays. Furthermore, attempts to obtain abnormal metaphases from involved bone marrow are often thwarted by the low mitotic index of the malignant cells and cellular contamination by non-malignant cells [307]; however, results from aCGH studies suggest that genomic imbalances occur in the neoplastic cells in most cases.
No unique recurrent abnormalities have been reported in ANKL, although conventional and molecular cytogenetic studies in this disorder are limited. Abnormalities common to other NK cell neoplasms are also found in ANKL, including del(6q), del(11q), del(13q), del(17p), trisomy 8, gain of the X chromosome, and isochromosomes of 1q, 6p, 7q and 17q, usually in karyotypes with a near-diploid modal number and 3 or more abnormalities [307–312]. Spectral karyotyping on a case of ANKL with a complex karyotype identified one translocation involving Xp21 and chromosome 15 together with an unbalanced t(1;8)(q10;p23), demonstrating translocation as a mechanism for gene deregulation in this condition. A translocation with an Xp21 and an 8p23 breakpoint respectively were also identified in an additional 2 cases of ENK/TL-NT analysed, suggesting that they may be recurrent breakpoints in these related NK cell disorders [313].
Siu et al. [314] performed conventional cytogenetics and metaphase CGH on patients with ANKL and ENK/TL-NT. The regions most commonly lost were 6q, 13q, 17p, 9p, 1p, 7p, 10p and 11q, and the most frequent gains were observed at 1p, 6p, 11q, 12q, 17q, 19p, 20q and Xp. Del(17p), del(11q) and gain of Xp were more often observed in ANKL than ENK/TL-NT. Del(6q), del(11q) and del(13q) have been considered the most frequent abnormalities in ANKL [309, 315]. However, a study of 22 Japanese patients confined purely to patients with ANKL failed to identify any instances of del(6q), suggesting that loco-regional differences in the frequency of cytogenetic abnormalities may also be at play [310]. More recently Nakashima et al. [316] performed aCGH on 10 ANKL and 17 ENK/TL-NT samples. In this series, gain of 1q, together with loss of 7p and 17p, was more likely to occur in the ANKL than in ENK/TL-NT. Notably, loss of 6q and 13q was again infrequent in ANKL in this Japanese study.
Adult T-Cell Leukaemia/Lymphoma
Adult T-cell leukaemia/lymphoma (ATLL) is a T-cell neoplasm encountered in the human T-cell lymphotrophic virus type 1 (HTLV-1) endemic areas of Japan, the Caribbean and Central Africa. ATLL is divided into four clinico-pathological subtypes: acute, lymphomatous, chronic and smouldering. Since chronic infection with HTLV-1 is the causative or initiating event leading to an expanded population of terminally differentiated CD4+, CD25+, CCR4+, FOXP3+ regulatory T cells, monoclonal integration of HTLV type 1 provirus into cellular DNA is the sine qua non of this disorder. Tumour clonality is readily demonstrable by other molecular means, and cytogenetic techniques are not considered necessary for optimal diagnosis and management of patients in routine clinical practice, but may be incorporated into the design of clinical trials as investigational tools [317].
The long latency between HTLV-1 infection in childhood event and disease onset (classically at 50 years of age or older) suggests that step-wise acquisition of additional genetic events is required for tumorigenesis. Accordingly, cytogenetic studies revealed karyotypes that were often complex with numerical and structural abnormalities. Frequent gain of chromosomes 3, 7 and 21 and loss of Y, 1p, 3q, 5q, 6q, 7p, 9q, 10p and 13q were seen by conventional karyotyping, but no abnormality was specific for ATLL [318–321]. Abnormality rates were high, with karyotypic abnormalities noted to be more frequent in aggressive than indolent disease [321]. In the study by Itoyama et al. [319], the presence of more than 6 unrelated abnormalities and abnormalities of 1p, 1q, 2q, 3q, 14q and 17q or loss of 2q, 9p and 17q was associated with shorter overall survival. Abnormalities at 14q11, the site of the TRA@/D@ locus, are reported in approximately 10–15 % of patients with ATLL [319, 321], and Haider et al. [322] demonstrated rearrangement of TRA@/D@ by FISH in one case of acute ATLL. A CGH study by Tsukasaki et al. [323] of 18 patients with indolent disease and 46 patients with aggressive disease found that genomic imbalance was more likely in aggressive ATLL subtypes. Gains of 14q, 7q and 3p were common; regions of loss were less frequent than gains, with the most commonly deleted regions being 6q and 13q; 14q and 7q gains were also reported in a smaller independent study by Ariyama et al. [324] involving ATLL cells lines and primary patient samples.
Oshiro et al. [325] compared BAC/PAC aCGH profiles from 17 cases of acute ATLL with 49 cases of lymphomatous ATLL. A greater number of genomic imbalances were apparent in the lymphomatous type. Recurrent regions of gain common to both types were 3p36, 3q25, 8q24, 9p24, 9q34 and 14q32. The most frequent regions of loss that were common to both types were 2q37, 4q21, 6q14, 9p21, 17p13.1 and 19q13. Gain of chromosome 3 or 3p was more common in acute ATLL, whereas gain of 7q and loss of 13q were observed more frequently in lymphomatous ATLL. Two-copy loss of 9p21 was frequent, consistent with homozygous deletion of CDKN2A and CDKN2B. It has been associated with disease progression from chronic to acute ATLL and with inferior clinical outcomes [326–328]. Deletions/mutations of TP53 at 17p13.1 have also been observed with disease progression and are estimated to occur in up to 40 % of cases of aggressive (acute or lymphomatous) ATLL [320, 329–331]. A recent study by Tawara et al. [332] of patients with newly diagnosed ATLL found a TP53 mutation rate of 17.9 % with similar rates in patients with acute (18.6 %) and chronic disease (15.4 %), whereas CDKN2A mutation was over-represented in the acute disease subtype relative to the chronic variant (44.2 % vs. 15.4 %). Strikingly, TP53 and CDKN2A mutations were mutually exclusive and patients lacking either mutation fared better in terms of overall survival. Furthermore, TP53 and CDNK2A mutation retained their prognostic significance in multivariate analysis.
Miyata et al. [333] performed BAC/PAC aCGH on DNA derived from skin lesions from patients with cutaneous disease. Gain of 1p, 7q, 18q and loss of 13q were recurrent findings (seen in greater than 20 % of cases). Gain of 14q32, observed in 44 % and 50 % of acute and lymphomatous subtypes of ATLL respectively [325, 334], was present in only 14 % of more indolent cutaneous cases. However, the study was not geared to determine if 14q32 gain tracked with increased rates of disease progression to more aggressive ATLL. Gain of 1p and loss of 13q were independent predictors of poor prognosis [333].
Extranodal NK/T-Cell Lymphoma Nasal Type
The more common ENK/TL-NT, a tumour involving the upper aerodigestive tract (“nasal” or NK/TL-N) or extranasal sites like skin, soft tissue, the gastrointestinal tract and testis (“nasal type” or NK/TL-NT), shares many features with ANKL, including a strong association with EBV infection and geographical distribution through Asia, Mexico, Central and South America. A CD2+, sCD3-, cytoplasmic CD3ε, CD56+, cytotoxic granule NK cell phenotype not dissimilar to the immunophenotype of ANKL is the norm, but occasional cases have a cytotoxic T-cell phenotype and a monoclonal TCR gene rearrangement.
The inaccessible anatomical location of the tumours and frequent necrosis make it challenging to obtain appropriate tumour material for conventional cytogenetic studies in many cases. Nevertheless there is a high abnormality rate. Recurrent cytogenetic abnormalities observed by conventional karyotyping include del(6q), del(13)q, del(17p), gain of the X chromosome, trisomy 8, isochromosomes of 1q, 6p, 7q and 17q and rearrangements involving Xp21, 8p23 and 11q23 [307, 311–313, 318, 335]. CGH studies by a Hong Kong-based group [314] of five cases of ENK/TL-NT revealed loss of X, 6q and 13q, and gain of 1p, 6p, 7q, 11q, 12q, 19, 20 and 22 in at least two cases. Using microsatellite markers in 15 patients, the same group showed LOH of 6q, 13q, 17p and 11q in 81 %, 67 %, 31 % and 29 % of cases respectively [315]. In 2001, Ko et al. [336] reported CGH studies in seven Korean patients with ENK/TL-NT and found gain of 2q, 13q, 10q, 21q, 3q, 5q and 17q to be most frequent; the regions most often lost were 1p, 17p, 12q and 13q. Notably only one patient in the Korean cohort [336] had deletion of 6q, an extremely frequent finding in the Hong Kong study. Indeed, overall there was very little overlap between the CGH findings of Siu et al. [315] and Ko et al. [336]; however, it is unclear whether this discrepancy between the two studies reflects small sample size, methodological differences, differences in histopathological classification or geographical differences in disease biology.
In 2005, Nakashima et al. [316] compared ten cases of ANKL with 17 cases of ENK/TL-NT by aCGH. Gain of 2q and loss of 6q16.1–q21, 11q22.3–q23.3, 5p14.1–14.3, 5q34–35.3, 1p36.23–p36.3, 2p16.1–16.3, 4q12 and 4q31.3–q32.1 were the strongest positive predictors for a diagnosis of ENK/TL-NT as opposed to a diagnosis of ANKL. Array CGH studies of seven patients with NK cell malignancy were reported by Iqbal et al. [337]. The pattern of genomic imbalances observed closely approximated the findings of Nakashima et al. [316] with some overlap with the dataset of Siu et al. [315], including gain of 1q, 2q, 6p, 7q, 17q and 20q and loss of 6q, 11q, 13q and 17p. Huang et al. [338] analysed nine cases of ENK/TL-NT by BAC aCGH. Recurrent areas of copy number gain were 1q21–q44, 4p16, 6p25–p11.1, 6q11.1–q14, 6q27, 7p11.2–q34, 7q11.2–q34, 7q35–q36, 8p23.3, 9q34, 10p15–p14, 11p15, 16p13.3, 17q12 and 22q11.21; recurrent areas of loss were 6q16–q25, 8p23–p22, 11q24–25 and 17p13.3. Interestingly, regional gain of 1q, del(6q) and del(17p) which were recurrent in this cohort were also found by Iqbal et al. [337]. Berti et al. [339] compared aCGH tumour profiles from three patients with primary cutaneous ENK/TL-NT to a skin tumour from one patient with disseminated ENK/TL-NT. Gain of 1q, 7q and deletion of 17p were seen in at least 2/3 patients with primary cutaneous disease and loss of 6q in 1 patient. The skin tumour from the patient with ENK/TL-NT also had gain of 7q along with loss of 9p and 12p that was not observed in the primary cutaneous tumours.
Enteropathy-Associated T-Cell Lymphoma
Enteropathy-associated T-cell lymphoma (EATL) is a highly aggressive primary T-cell neoplasm arising in the gastrointestinal tract. Monoclonal T-cell receptor gene rearrangements are present. Non-monomorphic or type I EATL is a disease of Western countries that has a strong association with coeliac disease. Tumour cells usually display surface expression of CD8 but not CD56 and have pleomorphic, anaplastic or immunoblastic morphology. In contrast EATL type II has a broader geographical distribution and an association with coeliac disease is not proven. Type II tumours have a monomorphic appearance and frequently co-express CD8 and CD56. Clonal cytogenetic abnormalities have been reported in only a small number of patients with EATL using conventional karyotyping, without any consistent pattern of derangement being identified [340].
In 2002, Zettl et al. [341] analysed DNA from 38 formalin-fixed paraffin-embedded tumours or fresh-frozen tumours by CGH. Copy number aberrations were present in 87 % of tumours studied. Strikingly gain of 9q was observed in 58 % of cases, with particularly localised gain at 9q33–34 confirmed by FISH testing. Other frequently gained regions were 7q (24 %), 5q (18 %) and 1q (16 %). 8p and 13q were deleted in 24 % of cases, 9p in 18 % and 16q in 8 %. Clinical outcomes were worse in patients with tumours that carried more than three copy number aberrations. Interestingly, partial trisomy of 1q has been identified in a high proportion (6/7) of patients with refractory coeliac sprue, a preneoplastic or precursor lesion to non-monomorphic or type I EATL, suggesting that deregulation of candidate genes on 1q is an early event in the transformation of intestinal T cells [342]. FISH studies by Leich et al. [343] in 2007 identified gain of 7q in 14/51 EATLs tested. As this included ten cases that had been studied by CGH previously [341], the FISH patterns could be interpreted in conjunction with imbalances detected by CGH. In at least two cases and up to six cases, the abnormalities detected were consistent with isochromosome formation as the mechanism for 7q gain in this disorder.
Deleeuw et al. [344] published the results of higher resolution aCGH studies on 30 samples in 2007, confirming and further refining the copy number imbalances detected by CGH. The 9q gain resolved into discrete regions of alteration at 9q31.3–q33.2, 9q33.2–q33.3 and 9q34.11-qter. There was copy number gain at 9q33.2–q33.3 in 67 % of cases. The 5q gain was mapped to 5q34–35.2. The 1q gain was reconfigured to three regions: 1q23.1–23.3, 1q25.3–31.2 and 1q32.2–41. The most distal region on 1q was most frequently involved (47 % of cases) and was highly amplified in one case. Gains on 7q were also found to be segmental and complex, with gain of 7q33–34 being the most common. In all, 36 % of cases had loss of 8p22–23.2, making this the most frequently lost region. The 16q deletion was mapped to 16q12.1. The 9p copy number loss spanned 9p21.2–p21.3, a region that encompasses the CDKN2A and CDKN2B genes. The investigators noted that 9q gain and 16q loss were almost mutually exclusive and one or other abnormality was present in virtually all case studies. Gain of the MYC oncogene at 8q24 was highly enriched for in samples from the monomorphic type II EATL patient cohort. Principal components analysis using all regions of alteration showed increased copy number at 8q24 and 1q gain were the major factors contributing to separation of non-monomorphic/type I EATL from type II EATL. In summary, gain of 9q31 or deletion of 16q21 occurs in greater than 80 % of cases of both EATL (type I) and type II EATL and appear to be unifying or characteristic of the condition as a whole. Furthermore, gain of 1q32.2–q41 (73 % vs. 20 %) and 5q34–35.2 (80 % vs. 20 %) is strongly over-represented in type I EATL compared to type II EATL, whereas gain of MYC at 8q24 is less frequent in type I EATL than in type II EATL (27 % vs. 73 %). A recent study reported aCGH results for 5 Korean patients with type II EATL. Gains of 9q33–q34, 6p21.21 and 19q21 and loss of 3q26.31 were observed in at least 2 cases. In keeping with the findings of Deleeuw et al., the frequency of 9q gain was 80 % and gain of 8q24 was seen in one patient [336].
Hepatosplenic T-Cell Lymphoma
Hepatosplenic T-cell lymphoma (HSTL) is a mature cytotoxic T-cell lymphoma involving the sinusoids of the spleen, liver and bone marrow that mainly affects young adults and children and can be seen in the setting of immunosuppression. Most tumours are derived from T cells of the γδ lineage, but rare cases that express TCR αβ heterodimers are also reported [345]. In cases where conventional cytogenetics has identified an abnormal clone, the modal number is most often in the near-diploid range and a mixture of abnormal and normal metaphases is usual. Isochromosome of 7q is the most common cytogenetic abnormality in HSTL [346–348]. Since a stemline with an isolated i(7q) is often encountered together with sidelines containing additional cytogenetic abnormalities, the i(7q) is thought to be the primary cytogenetic abnormality in this condition. More recently variants resulting in gain of 7q as a result of ring chromosome formation have been reported [349, 350]. It is unclear if loss of 7p material or gain of 7q material is of greater consequence in i(7q) formation in HSTL. However, rare cases that demonstrate gain of 7q without coincident 7p loss, or gain of additional copies of 7q with disease progression favour gain of genetic material on 7q as the event most likely to confer a selective advantage on the neoplastic clone [349, 351, 352].
In most reported HSTL cases with i(7q), the karyotypes are relatively simple with no more than three abnormalities. Trisomy 8 is the most common secondary abnormality described, followed by loss of the Y chromosome [346–350]. Isochromosome of 7q is not specific for HSTL and has also been reported in other mature T-cell neoplasms, T-cell and B-cell acute lymphoblastic leukaemia, acute myeloid leukaemia, myelodysplastic syndromes and Wilm tumour. The difference is that in conditions other than HSTL, the i(7q) is usually present in complex karyotypes, as a secondary abnormality or seen to arise during clonal evolution with disease progression. Thus, the combination of i(7q) with trisomy 8 in a relatively simple karytoype is most characteristic of HSTL.
However, HSTL cases without i(7q) also exist, including cases with increased complexity [353]. A case series investigating the frequency of TCR translocation events and isochromosome 7q formation in T-cell lymphomas did not identify any abnormalities in four cases of HSTL [335], although at least three of these cases were confirmed to be of the αβ type in which i(7q) is infrequent [352]. An early study had suggested that i(7q) was an adverse prognostic factor in HSTL [354], but this was not borne out in retrospective analysis of a larger case series [353].
Mycosis Fungoides/Sézary Syndrome
Primary cutaneous T-cell lymphomas (CTCL) represent a group of malignancies of mature T cells that manifest primarily in the skin. The most common CTCL is mycosis fungoides (MF) which presents with skin lesions containing clonal malignant T cells, but CTCL may also present in a leukaemic form with skin involvement and lymphadenopathy, Sezary syndrome (SS). SS is an aggressive form of CTCL defined by the triad of erythroderma, generalised lymphadenopathy and the presence of neoplastic T cells in skin, lymph node and peripheral blood. Approximately 10 %–20 % MF transforms to a large T-cell lymphoma over time.
Conventional cytogenetic studies of CTCL have been hampered by the difficulty in producing good quality metaphase spreads from malignant cutaneous lymphocytes which may be mixed with a large number of reactive lymphocytes. In consequence, low-grade CTCL have appeared more likely to have a normal karyotype. In contrast, high-grade CTCL have been reported to have deletions of 6q and 13q and trisomy of 7q; these abnormalities have been shown to involve deletion of 6q21, amplification of 7q21 and interstitial deletion of 13q14 [355].
A small study by Thangavelu et al. of 19 patients with SS/MF identified clonal abnormalities by conventional cytogenetics in 53 % of cases [356]. The commonest abnormalities were deletions of 1p22–1p36, rearrangements involving chromosomes 10 and 17 and, in two cases, a der(8)t(8;17)(p11;q11) was seen. Regions of the genome encoding T-cell receptors were not involved in abnormalities.
Espinet et al. [357] studied another 21 patients with SS using both conventional cytogenetics and cross-species colour banding and found the most common structural abnormalities to involve 1q, 2q, 6q and 8q and the most common numerical abnormalities to be monosomies of 9 and 10 and trisomy 18 with monosomy 10 in 73 % of abnormal cases. Multicolour FISH applied to 9 abnormal MF/SS karyotypes also showed abnormalities of 10q to be the most common, observed in 7/9 cases [358]. Recurrent breakpoints were 1p32–36, 6q22–25, 17p11.2–13, 10q23–26 and 19p13.3, and one patient also showed the dicentric 8;17 translocation described previously in two patients with SS by Thangavelu et al. [356]. Loss of heterozygosity was examined at a number of loci in 19 MF patients and frequent LOH confirmed at 10q23 (PTEN) and 17p13 (TP53) [359].
Array data from a number of publications are summarised in Table 44.3. The most frequently reported copy number gains are of 17q and, in two series of SS cases, gain of 8q encompassing MYC was observed. The most frequent recurrent losses were of 10q and 17p.
Table 44.3
Array data showing recurrent copy number gains and losses in MF and SS
Dx (no. studied) | Gains | Losses | References |
|---|---|---|---|
MF (22) | 1p36.2, 1q31–1q32, multiple regions on 7q and 7p | 5q13, 9p21, 13q14–13q31 | [360] |
MF (16), SS (18) | 4/4q, 17q/17, 18 | 1p31–36, 10q26, 17p, 19 | [429] |
MF (3), SS (4) | 17q11.2–17q12a | 10q23 | [364] |
MF (15), SS (11) | 10q23.33–24.1, 10q24.33–10q25.1 | [363] | |
MF-T (11)b | 1p36, 7, 9q34, 17q24-qter, 19 | 2q36-qter, 9p21, 17p | [430] |
SS (20) | 2p11.2, 4p16.1, 7p21.1, 8q24.1–q24.3, 17q21.31–q23 | 3q26.33, 5q14.3, 7p14.1, 9p13.1–9p2, multiple regions on 10, 16p11.2, 17p13–17p12 | [361] |
SS (28) | 17p11.2–17q25.3, 8/8q, 10p15.3–10p12.2 | 10p12.1–10q16.3, 17p13.2–17p11.2, 9q13–q21.33 | [431] |
SS (6) | 22q11.2–22q13.3 | 1p36–1p22, 6q24, 15q11.2 | [432] |
Comparing aCGH studies of 22 patients with tumour-stage MF and 20 SS patients did not identify much overlap in the recurrent chromosome aneuploides seen in the two groups [360, 361]. Five SS patients and 2 MF patients were studied using a genomic microarray containing 57 oncogenes and showed oncogene copy number gains of RAF1 (3p25), CTSB (8p22), PAK1 (11q13) and JUNB (19p13) in 5/7 patients. A further 23 SS and 10 MF patients were subsequently studied by immunohistochemistry and showed expression of nuclear JUNB in 21/23 SS and 5/10 MF patients [362].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


