AML with t(8;21)(q22;q22)/RUNX1-RUNX1T1 (AML1-ETO)
AML with inv(16)(p13.1q22) or t(16;16)(p13.1q22)/CBFB-MYH11
APL with t(15;17)(q22;q21)/PML-RARA
AML with t(9;11)(p22;q23)/MLLT3 -MLL (specify other 11q23/MLL abnormality)
AML with t(6;9)(p23;q34)/DEK-NUP214(CAN)a
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2)/RPN1-MECOM a
AML with t(1;22)(p13;q13)/RBM15-MKL1(OTT-MAL)a
Provisional entity: AML with mutated NPM1 a
Provisional entity: AML with mutated CEBPA a
Favorable prognostic factors for patients with AML include low white blood cell (WBC) count; inv(16)/t(16;16); t(8;21); t(15;17); Down Syndrome (DS) in children up to 4 years old, mostly with acute megakaryoblastic leukemia (AMkL, AML-M7); and early response to treatment. Unfavorable prognostic factors include -7, -5/5q-, t(6;9), t(6;11), 3q abnormalities, complex karyotypes (≥3 cytogenetic findings, excluding the categories described by WHO-2008), and FLT3-ITD, particularly with a high allelic ratio. Intermediate-risk factors include normal and other karyotypes, excluding favorable and unfavorable abnormalities [1].
AML represents 15–20 % of all childhood leukemias and approximately 33 % of adolescent and 50 % of adult leukemias. The prognostic value of cytogenetics has been well established for all age groups [9]. However, the distribution of recurrent chromosomal abnormalities observed in AML differs among various age groups. In infants (<2 years old), chromosomal abnormalities most frequently have a breakpoint in 11q23 and involve MLL in 60 % of cases. The incidence decreases to 30 % in children (2 to <13 years) and to 13 % in adolescents (13 to <21 years), and is even lower in young adults (8 %; 21 to <30 years) and adults (3 %; ≥30 years) [9]. The t(1;22)(p13;q13) is specific for AMkL and is mostly seen in children younger than 2 years and rarely in adults. In older children (>10 years) and young adults (<60 years), the most frequent abnormalities are t(15;17), t(8;21), and inv(16). In older patients (>60 years) with AML, there is a low frequency of favorable core-binding chromosomal abnormalities but a higher incidence of complex aberrant karyotypes, which is attributed to both disease biology and host factors [10]. A recent study evaluating the impact of age on outcome of AML in children 21 years or younger has suggested that age is an independent prognostic factor in childhood AML and that children younger than 10 years benefit more than older children from newer intensive therapies [11].
The following section describes the associations among specific cytogenetic and molecular changes in AML, distinct clinical subtypes of diseases, and treatment outcomes.
Specific Recurring Cytogenetic and Molecular Abnormalities
t(15;17)(q22;q12)/PML-RARA
The t(15;17) is associated with APL, a distinct subtype of AML that is treated differently from other types of AML because of its marked sensitivity to the differentiating effects of all-trans retinoic acid (ATRA). The t(15;17) is found in 9 % of children with AML, it is rarely observed in infants, and the incidence decreases with age in adults (rarely seen in adults 45 years or older) (Fig. 18.1). The t(15;17) generates the fusion gene PML-RARA to produce a chimeric protein. Patients with the t(15;17) and APL respond well to the combination therapy of ATRA and the chemotherapy agent arsenic trioxide (ATO) and have a favorable prognosis (cure rate 80 %) [12–14].
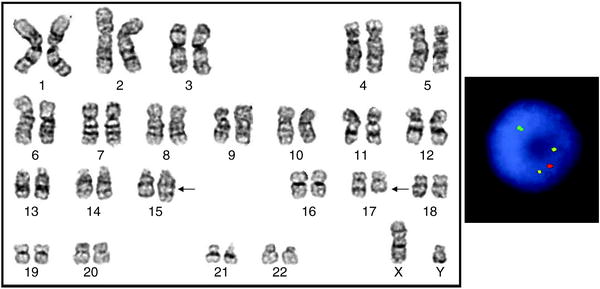
Fig. 18.1
Left: Karyogram of a cell from a patient with APL and the typical translocation between chromosomes 15 and 17 (arrows). The karyotype is 46,XY,t(15;17)(q22;q21). Arrows indicate abnormal chromosomes. Right: FISH with the dual-color dual-fusion PML/RARA probes (Abbott Molecular) showing 2 fusions (yellow), 1 red, and 1 green signal
Other rare translocations involving RARA can also result in APL. Approximately 1 % of patients with APL have variant translocations involving only RARA. The most frequent is t(11;17)(q23;q21)/PLZF-RARA, and patients with this translocation are partially resistant to ATRA. Other variant translocations with RARA include NPM1, STAT5B, and NUMA1 (Table 18.2). It is important to identify such patients, as the sensitivity for ATRA or ATO may vary [15, 16].
Table 18.2
Variant RARA aberrations in APL and its response to ATRA
Chromosome abnormality | Partner breakpoint | Gene | ATRA response |
|---|---|---|---|
t(15;17)(q22;q21) | 15q22 | PML | Yes |
t(5;17)(q35;q21) | 5q35 (n = 5)a | NPM1 | Yes |
t(11;17)(q23;q21) | 11q23 (n = 20) | PLZF(ZBTB16) | No |
der(17)ins(17)(q21q11.2) | 17q11.2 (n = 1) | STAT5B | No |
dup(17)(q21.3q23) | |||
ins(17;17)(q21q24) | 17q24 (n = 1) | PRKAR1A | Yes |
t(4;17)(q12;q21) | 4q12 (n = 1) | FIP1L1 | Yes |
t(11;17)(q13;q21) | 11q13 (n = 1) | NUMA1 | Yes |
A suspected diagnosis of APL should be treated as a medical emergency because immediate measures are needed to counteract coagulopathy and initiate ATRA therapy. Thus, a prompt genetic diagnosis by conventional cytogenetics, FISH with PML–RARA probes, RT-PCR, or anti-PML monoclonal antibodies, is essential for patients with APL [13]. Conventional G-banding has helped to identify the t(15;17) in more than 90 % of patients with APL, but it cannot detect the PML-RARA fusion resulting from cryptic rearrangements. However, cytogenetics facilitates the identification of rare variant translocations, including three-way translocations affecting PML and RARA or other aberrations affecting only RARA. FISH with commercially available probes is a rapid, highly sensitive, and specific method to confirm the presence of the PML-RARA fusion. However, in rare instances, fusion signals may be different from those expected, because of deletion of sequences at translocation junctions or where PML-RARA is formed as a result of an insertion or a complex translocation [4]. FISH using RARA breakapart probes is useful to evaluate rearrangements in suspected cases that lack the PML-RARA fusion. RT-PCR is important to establish the type of PML-RARA isoform fusion transcript that will be the target to reliably monitor minimal residual disease. Immunostaining with anti-PML monoclonal antibodies on dry smears is also useful to establish a rapid diagnosis.
Small PML-RARA insertions can be missed by FISH when using very large probes; in such cases, it is more appropriate to use relatively small cosmid probes. Patients with APL in whom the PML-RARA fusion is generated due to variant three-way translocations or cryptic insertions respond to ATRA combination regimens in a similar manner as those with APL with the typical t(15;17) [4].
Children with APL have a higher incidence of hyperleukocytosis (WBC count ≥10 × 109/L) than adults with APL, but the high WBC count is associated with poor outcome in both groups [12]. FLT3 mutations [either internal tandem duplications (ITDs) or kinase domain mutations] have been observed in up to 40 % of patients with APL and are associated with high WBC counts; however, there is no clear correlation between the presence of FLT3 mutations and outcome [17].
Core-Binding Factor Leukemias
In children and adults with AML, the highest complete response rates and longest survival times are associated with the two common translocations t(8;21) and either inv(16) or t(16;16). Both translocations disrupt the core-binding protein complex, which plays a vital role in hematopoiesis, and are called core-binding factor (CBF) leukemias. The t(8;21) targets RUNX1 and the inv(16)/t(16;16) targets CBFB.
t(8;21)(q22;q22)/RUNX1(AML1,CBFA2)-RUNX1T1(ETO,MTG8)
The t(8;21)(q22;q22) is among the most frequent recurrent abnormalities in AML (12 % of children and 6 % of adults). The t(8;21) juxtaposes RUNX1 to RUNX1T1 on the derivative chromosome 8, generating a chimeric gene RUNX1-RUNX1T1 (AML1-ETO), which inhibits transcriptional activation by wild-type RUNX1. RT-PCR and RUNX1-RUNX1T1 FISH probes are available to detect the t(8;21), and are useful especially when submicroscopic rearrangements are suspected or the sample does not yield metaphases for analysis. The most frequent additional change in cases with t(8;21) is loss of a sex chromosome in approximately 40 % of cases; the Y chromosome is lost in males more frequently (55 %) than the X chromosome (33 %) in females [18]. A del(9q) or rearrangement of 7q can also occur, but this does not significantly affect overall survival. The t(8;21) is associated with a favorable outcome, especially when the disease is treated with regimens containing high-dose cytarabine [19].
Presence of the RUNX1–RUNX1T1 transcript in the bone marrow in adults and cord blood cells of children who do not have AML suggests that the t(8;21) is generated early in hematopoiesis. Some people may acquire secondary genetic alterations that lead to the development of AML [20], which is supported by the detection of RUNX1–RUNX1T1 in neonatal blood spots of infants in whom AML subsequently developed (after >10 years) [21].
inv(16)(p13.1q22) and t(16;16)(p13.1;q22)/CBFB-MYH11 (SMMHC)
The inv(16) and t(16;16) result in fusion of CBFB at 16q22 to the smooth muscle myosin heavy chain gene (MYH11) at 16p13.1. CBFB is the heterodimeric partner of RUNX1, which together form the transcriptional activating factor designated as CBF. The inv(16)/t(16;16) is strongly associated with the FAB M4Eo subtype and is observed in approximately 6 % of children and adults with AML. FISH probes and RT-PCR are available to evaluate the status of fusion genes in patients with AML and can aid the diagnosis of this subtle chromosomal abnormality; trisomy of 22 is a frequent additional abnormality in leukemic cells with an inv(16). The inv(16)/t(16;16) confers a good prognosis in both children and adults with AML.
Other RUNX1(AML1,CBFA2) (21q22) Rearrangements
The RUNX1gene encodes a transcription factor important for hematopoiesis and is one of the most frequently mutated genes in acute leukemias. Mutations include the t(12;21)(p13;q22)/ETV6-RUNX1(TEL-AML1) in ALL and many other RUNX1 partner genes identified in AML, but several of them are reported for single cases.
One of the most frequently identified RUNX1 partner genes is MECOM involved in the t(3;21)(q26;q22) associated with therapy-related MDS and CML in blast crisis [22]. The t(3;21) can generate fusion of RUNX1 with several genes such as EVI1, EAP, and MDS1. In addition, other rearrangements of 3q26 [such as the inv(3)(q21q26)] often lead to overexpression of EVI1, which is associated with poor prognosis, suggesting its role in the pathogenesis of AML [23]. Determination of EVI1 expression is likely important, because it is overexpressed in cases with cryptic 3q26 rearrangements [24]. Another rare but recurrent translocation is the t(16;21)(q24;q22), mostly observed in patients with therapy-related acute leukemias and MDS but also in children and adults with de novo AML. The t(16;21) generates the fusion gene RUNX1-CBFA2T (MTG16), which is similar to the RUNX1–RUNX1T1 generated by the t(8;21).
q23/MLL (MLL1,ALL1,TRX,HTRX1)
Most myeloid blasts with an 11q23 abnormality have an MLL rearrangement and are associated with monocytic differentiation. Every MLL translocation results in gain of function by generating novel chimeric proteins coded by the N terminus of MLL fused in-frame with one of many partner genes coding for diverse functions, and each oncogene is associated with a distinct form of leukemia [25]. More than 100 translocations target MLL in acute leukemias, and approximately 64 partner genes for MLL have been cloned [6]. Despite the diversity of chimeric proteins created, MLL fusions generate a characteristic gene expression profile (GEP), including upregulation of developmental homeobox genes such as HOXA9, suggesting that partner genes for MLL may be involved in regulating transcription elongation and chromatin structure remodeling.
Chromosomal rearrangements involving MLL in AML and in ALL include balanced and unbalanced translocations, inversions, insertions, amplification, and partial tandem duplication (PTD). These genetic mechanisms convert MLL into a chimeric transcription factor with leukemogenic properties [6]. Some MLL gene rearrangements are not detected by conventional cytogenetic methods. FISH using commercially available dual-color MLL probes allows evaluation of derivatives of a translocation involving MLL in metaphase chromosomes and the splitting of signals in interphase nuclei (Fig. 18.2). In rare instances, FISH using this probe detects not only the reciprocal translocation but also a deletion of at least 190 kb from the 3′ region of MLL.
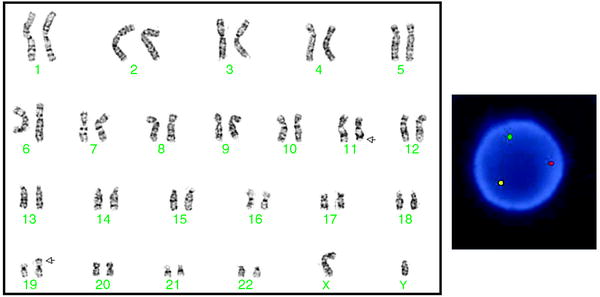
Fig. 18.2
Left: Karyogram of a cell with a t(11;19)(q23;p13.3), which occurs in both AML and T-cell ALL. Arrows indicate abnormal chromosomes. The karyotype is 46,XY,t(11;19)(q23;p13.3). Right: FISH with an MLL breakapart probe (Abbott Molecular) shows the separation of the 2 MLL signals (1 red and 1 green) and a normal fused (yellow) signal indicating a rearrangement of the MLL gene
The specific fusion partner of MLL may influence prognosis. For example, the t(4;11)(q21;q23) is one of the most frequent rearrangements of 11q23 in ALL reported primarily in infants and young children (70 % 0–2 years old; 50 % under 4 years) and is usually associated with an unfavorable prognosis; the rearrangement has been occasionally observed in adults and in myeloid leukemias too. Contrary to the poor prognosis of infants with ALL and 11q23/MLL +, the outcome of infants with 11q23/MLL + AML is not poorer than that of infants who lack this rearrangement. Several studies have reported a more favorable prognosis for children with t(9;11)(p22;q23)/MLL-MLLT3, but other studies have refuted these observations. WHO-2008 has classified the t(9;11) as a separate category and recommends that partner genes be determined for cases with MLL rearrangements [1].
In a recent collaborative group study of 756 children with AML and an 11q23/MLL rearrangement, the overall 5-year event-free survival (EFS) rate was 44 %. The outcome of the subgroups varied greatly; for example, patients with a t(1;11)(q21;q23) had an excellent outcome (5-year EFS, 92 %), whereas those with a t(6;11)(q27;q23) had the worst outcome (5-year EFS, 11 %). Subgroups t(10;11)(p12;q23), t(4;11)(q21;q23), and t(10;11)(p11.2;q23) had a 5-year EFS of 31 %, 29 %, and 17 %, respectively [26].
An MLL-PTD is detected in approximately 10 % of patients with AML, particularly those with trisomy 11 or a normal karyotype, and confers a poor prognosis [27]. In the 1990s, the use of epipodophyllotoxins was associated with the development of therapy-related AML (t-AML) generally having 11q23 abnormalities. The most frequently observed translocations were t(9;11)(p22;q23), t(11;19)(q23;p13.3), and t(11;16)(q23;p13.3). The last translocation, affecting CREBBP(CBP), is rarely seen in de novo AML. Subsequently, the dose and schedule of topoisomerase II inhibitors for treatment of acute leukemias were modified, which has reduced the occurrence of t-AML with an MLL rearrangement.
t(10;11)(p12;q14)/PICALM(CALM)-MLLT10(AF10)
This is a rare recurrent abnormality seen in both adult and pediatric patients with AML or T-ALL and is often associated with a poor prognosis [28, 29]. The t(10;11)(p12;q14) lacks an MLL rearrangement and may be difficult to differentiate microscopically from the variant t(10;11)(p12;q23)/MLL-MLLT10, and FISH or RT-PCR analysis is usually required to distinguish these two translocations with accuracy.
t(1;22)(p13;q13)/RBM15(OTT)-MKL1(MAL)
The t(1;22)(p13;q13) is restricted to patients with AMkL and occurs in 33 % of this subgroup, mostly in infants and young children. The t(1;22) is rare in children with DS who develop AMkL. The t(1;22) juxtaposes the OTT(RBM15) on 1p13 to MAL(MLK1) on 22q13. Patients with this translocation were earlier considered a high-risk category, but the use of intensive therapy has significantly improved prognosis [30].
t(6;9)(p23;q34)
Recurrent Abnormalities of Chromosomes 3, 5, and 7
Cytogenetic aberrations associated with unfavorable outcomes include monosomy 5 (−5) and del(5q)/5q-, monosomy 7 (−7), abnormalities of 3q [inv(3)(q21q26) or t(3;3)(q21;q26)], and complex abnormalities. These abnormalities are seen more frequently in adults than children. Patients with del(7q)/7q- are no longer considered to be at high risk of treatment failure [33].
t(8;16)(p11.1;p13.3)/MYST3(MOZ/KAT6A)-CREBBP(CBP)
Disruption of MYST3 at 8p11.1 is rare and is seen in patients with acute myelomonocytic or monocytic leukemia and t-AML, in which the leukemic blast cells frequently display erythrophagocytosis; alteration in MYST3 is associated with a poor prognosis [34]. In AML blast cells, the most frequent translocation is t(8;16)(p11.1;p13.3), which fuses MYST3 to CREBB(CBP). Other MYST3 fusion partners are NCOA2(TIF2) in inv(8)(p11.1q13), NCOA3 in t(8;20)(p11.1;q12), and P300(EP300) in t(8;22)(p11.1;q13) [34, 35]. These partner genes encode proteins that have histone acetyltransferase activity and likely contribute to leukemogenesis by altering chromatin-mediated transcriptional control of unknown target genes.
Recent GEP studies comparing adult patients with t(8;16) AML to those with normal karyotypes or other recurrent translocations have shown that those with t(8;16) were clustered close to 11q23/MLL cases and shared commonly expressed genes. On this basis, t(8;16) has been defined as a highly unique signature for AML [36–38]. This suggests that blasts in t(8;16) AML may originate from an early stem cell with myeloid and monoblastic characteristics, which may explain in part the unfavorable response of patients with t(8;16) AML to standard chemotherapy regimens. The CREBBP(CBP) (16p13.3) gene is also involved in other translocations in AML—t(10;16)(q22;p13.3)/MYST4(MORF)-CREBBP and t(11;16)(q23;p13.3)—which results in MLL-CREBBP(CBP) fusion and is strongly associated with treatment-related hematologic malignancies [39].
Complex Karyotype
The definition of a complex karyotype has not yet been standardized, but the current trend is to consider patients with three or more chromosomal abnormalities (excluding those recognized as categories by WHO-2008) in this category; adults with AML who have complex karyotypes have a significantly poorer prognosis than those without a complex karyotype [10]. The loss of 17p is among the most frequent abnormalities observed as part of a complex karyotype in hematologic disorders in adults, and is strongly associated with abnormalities of chromosomes 5 or 7, or both [40]. The loss of 17p consistently creates a monoallelic loss of TP53 and may be associated with TP53 mutation or lack of expression, which is associated with a very poor prognosis; risk-adapted targeted therapies are being evaluated for patients in this category [41].
Myeloid Proliferations Related to DS with AML
Transient Abnormal Myelopoiesis
DS with transient myeloproliferative disorder (TMD) is observed in infants with DS and is characterized by a clonal expansion of myeloblasts that can be difficult to distinguish from that seen in AML. Most cases of TMD regress spontaneously within the first 3 months of life. TMD blasts are most commonly megakaryoblastic and have distinctive mutations involving GATA1. TMD may occur in phenotypically normal infants with genetic mosaicism in the bone marrow for trisomy 21. Although patients with TMD rarely have other chromosomal abnormalities in addition to the +21, the presence of an aberration may predict an increased risk for developing AML [42]. Approximately 20 % of infants with DS and TMD eventually develop AML, most being diagnosed within the first 3 years of life [43].
Myeloid Leukemia Associated with DS
In general, the outcome of patients with DS who develop AML is favorable. Children younger than 4 years, who comprise the majority of patients with DS having AML, have an EFS greater than 80 % [42, 44, 45]. A distinctive feature of myeloid leukemias associated with DS is the presence of somatic mutations in the gene encoding factor GATA1. GATA1 (Xp11.23) mutations are present in most DS patients with either TMD or AMkL and appear to confer increased sensitivity to regimens containing cytarabine [46–48].
Genetic Mutations and Cytogenetic Subgroups
FLT3(13q12) Mutation
FLT3, a receptor tyrosine kinase expressed on hematopoietic progenitors, is frequently mutated in AML, mainly in patients with a normal karyotype [49]. The majority of FLT3 mutations are ITDs that generate in-frame insertions within the juxtamembrane region of the receptor. The presence of an FLT3-ITD has been associated with poor prognosis in both children (12 %) and adults (30 %) with AML; it is rare in children younger than 10 years. The poor prognosis is particularly significant when both alleles are mutated or there is a high ratio of the mutant allele to the normal allele [50, 51]. In addition to the FLT3-ITD, less frequent mutations involving the region encoding the activation loop (tyrosine kinase domain, TKD) have been identified in less than 10 % of children and adults with AML, but the clinical significance of these mutations has not been clearly defined [52]. FLT3-ITD and point mutations are particularly prevalent in APL (up to 40 % of children and adults), but their prognostic significance is unknown [53].
Isolated trisomy 13 is extremely rare in children, but has been reported in a low incidence in adults with AML or MDS. Such patients have a poor outcome; however, recent reports have shown that these patients may be sensitive to lenalidomide as are patients with low-risk MDS with the 5q-cytogenetic abnormality [54]. Trisomy 13 (chromosome 13 harbors FLT3) is strongly associated with RUNX1 (AML1) mutations and increased FLT3 expression in AML.
NPM1 (Nucleophosmin)(5q31) Mutation
The NPM1 protein has diverse functions in the cell, such as chromatin remodeling and ribosomal complex assembly. NPM1 mutations are detected by immunohistochemical methods that show its cytoplasmic localization [55]. NPM1 mutations are found in up to 35 % of adults and 8 % of children with AML, mostly in those with a normal karyotype, and are associated with good prognosis if FLT3 mutations are not present [51, 56].
CEBPA (19q13.1) Mutations
CEBPA, encoding a transcription factor involved in normal myelopoiesis, is mutated in 10 % of adults younger than 60 years and in 5 % of children with AML [57]. Recent reports have suggested that biallelic mutations of CEBPA associated with a normal karyotype predict a favorable outcome in the absence of FLT3 mutations [58–60].
RAS and KIT (4q12) Mutations
RAS mutations are present in 15 % of adults with AML, and these patients may have an increased sensitivity to cytarabine and benefit from higher cytarabine doses than those administered to patients with wild-type RAS [61]. KIT mutations are observed in 5 % of adults with AML and in up to 40 % of patients in the CBF subgroup and appear to be associated with a poorer outcome than that of patients with CBF AML without a KIT mutation [62, 63].
Summary
Conventional and molecular cytogenetics are among the most important features currently used to direct treatment randomization in AML. In the last 20 years, many recurrent genetic lesions have been identified, but no single mutation has been shown to be sufficient to cause leukemogenesis. Instead, cooperating mutations are believed to promote proliferation and survival of aberrant cells or block the differentiation necessary for leukemic transformation. Most of the recurrent chromosomal abnormalities identified in myeloid blast cells generate chimeric oncoproteins that inhibit differentiation. Recent molecular genetic studies have shown that the most common molecular markers for subclasses of AML include genetic lesions that may or not be associated with chromosome abnormalities as determined by conventional cytogenetics [66–68]. The advent of novel technologies available for the genomic sequencing of leukemic blasts will help enhance our understanding of the pathophysiology of acute leukemias and identify novel therapeutic targets.
Acute Lymphoblastic Leukemia
ALL is the most common leukemia in children, where 80–85 % of leukemias are ALL, and is much rarer in adults where only 20 % of acute leukemia is ALL. The cytogenetics of ALL has long been known to be of prognostic relevance, and both the number of chromosomes (modal number) and some structural abnormalities are prognostic in both adult and pediatric ALL [69].
Both cytogenetic aberrations and outcome differ between children and adults with ALL. Children with ALL have a very good prognosis, with 5-year EFS rates of more than 80 %. On the other hand, adults with ALL have a much poorer outcome with an overall EFS of 40 % [70]. This difference in outcome can in part be attributed to the different biology of ALL in the two age groups, some of which is demonstrated by cytogenetics. The frequencies of “good prognosis” cytogenetic aberrations are higher in children than in adults with ALL, and “poor prognosis” cytogenetic abnormalities are more frequent in adults than in children with ALL.
ALL also is differentiated by the cell type that becomes leukemic. Approximately 85 % of children and 75–80 % of adults with ALL have B-cell precursor (BCP) ALL, and the remaining have T-cell ALL (T-ALL) [71, 72]. The cytogenetics of and BCP and T-ALL differ, although there are some overlaps, and will be discussed separately. (See Table 18.3 for summary of ALL cytogenetic aberrations.)
Table 18.3
Common recurring aberrations in ALL
B-cell precursor ALL | ||||||
|---|---|---|---|---|---|---|
Frequency (%) | ||||||
Aberration | Adult | Pediatric | Genes | Prognosis | Cytogenetic variants | Comments |
Numerical aberrations | ||||||
Hyperdiploidy, mn > 50 | <10 | 25–30 | Good | |||
Hyperdiploidy, mn > 46 < 51 | 15 | 15 | Intermediate | Usually have structural abnormalities, ∼50 % recurrent | ||
Pseudodiploidy | Intermediate | |||||
Near-triploidy and mn 30–39 | Rare | Poor | ||||
Hypodiploidy, mn 44–45 | 7 | Intermediate | Usually have structural abnormalities | |||
Hypodiploidy, mn < 44 | 1.7 | Poor | Few structural abnormalities, frequent doubling in mn < 40 | |||
Structural aberrations | ||||||
t(9;22)(q34.1;q11.2) | 25 | <4 | BCR-ABL1 | Poor | 5 % complex | Frequency increases with age |
t(12;21)(p13;q22) | Very rare | 25 | ETV6-RUNX1 | Good | Cytogenetically cryptic. Many other ETV6 rearrangements, may differ in clinical characteristics and prognosis | |
t(4;11)(q21.3;q23) | MLL-AFF1 | Poor | Insertions, three-way translocations | Frequent in infants. Outcome may differ with age | ||
t(11;19)(q23;p13.3) | MLL-MLLT1 | Poor | Also in T-ALL | |||
t(9;11)(p22;q23) | MLL-MLLT3 | Adults better than other 11q23; children poor | ||||
Other 11q23 | 5 | 5 > 1 year 80 < 1 year | MLL/other | Poor | Frequent in infants. Outcome may differ with age and partner | |
t(1;19)(q23.3;p13.3) | 3 | 6 | TCF3-PBX1 | Not prognostic | der(19)t(1;19)(q23.3;p13.3); t(17;19)(q22;p13.3) | HLF-E2A in t(17;19)—poor prognosis |
der(21)/add(21) | RUNX1 | Poor | der(21) appears in many guises | Amplification of RUNX1 or near gene | ||
del(9p) | 30 | 6 | CDKN2A | Unknown | ||
dic(9;12)(p13;p11.2) | Rare | Rare | 50 % PAX5-ETV6 | Good | Not all have PAX5/ETV6 rearrangement | |
dic(9;20)(p12-p13;q11.2) | Rare | Rare | Some PAX5-ASXL1 | Good | ||
dic(7;9)(p11.2;p13) | Rare | Rare | ||||
t(8;14)(q24;q32) | IGH@-MYC | Usually Burkitt leukemia/lymphoma | ||||
t(8;14)(q11.2;q32) | IGH@-CEBPD | Good in Down syndrome | Prevalent in Down syndrome ALL | |||
t(5;14)(q31;q32) | Rare | Rare | IGH@-IL3 | Poor | Associated with hypereosinophilia | |
other 14q32 | IGH@ | |||||
del(6q) | 7 | 1.5 | MYB some cases | Not prognostic | Frequent in ETV6-RUNX1 cases | |
del(5q) | Rare | Rare | ||||
del(7p) | 4 | 4 | Poor | |||
del(12)(p11.2-p13) | ?ETV6 | Not prognostic | ||||
Sole +8 | 3 | 3 | Unknown | |||
Sole +X | Rare | Rare | Not prognostic | |||
Sole +21 | Rare | Rare | ||||
del(13q) monosomy 13 | 5 | 2 | Unknown | |||
T-lineage ALL a | ||||||
14q11.2 rearrangement | 35 | TRA@/TRD@ | Not prognostic | 7p14, 7q34 | TRG@,TRB@ | |
t(1;14)(p32;q11.2) | 10 translocation 30 deletion | TRA@/TRD@-TAL1 | Favorable | del(1)(p32p32) cryptic | TAL1-STIL | |
t(11;14)(p13;q11.2) | 5–10 | TRA@/TRD@-LMO2 | ||||
t(11;14)(p15;q11.2) | 2 | TRA@/TRD@-LMO1 | ||||
t(10;14)(q24.32;q11.2) | 5–10 | TRA@/TRD@-TLX1 | Favorable | |||
inv(7)(p15.2q34) | 3–5 | TRB@-HOXA@ | ||||
t(6;7)(q23;q34) | 3 | TRA@/TRD@-MYB | ||||
t(6;11)(q27;q23) | Rare
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||||