Non-hyperdiploid MM
Hyperdiploid MM
Translocations
Less translocations
Younger
Older
IgA
IgG
Lambda
Kappa
Less bone disease
More bone disease
More aggressive
More indolent
Females
Males
Chromosome 13 deleted
Chromosome 13 normal
Cytogenetic Studies in Interphase Cells
Fluorescent in situ hybridization (FISH) can be used to analyze either metaphase chromosomes or interphase cells (Fig. 30.1). Using FISH we now know that all PC dyscrasias (including MGUS) harbor both numerical and structural chromosomal abnormalities [23–25]. The incorporation of simultaneous fluorescent staining of cytoplasmic immunoglobulins and chromosomal DNA (cIg-FISH) [26] allows for the precise identification of clonally restricted abnormalities. This is especially important in cases with a low percentage of MM tumor cells since the malignant PC coexists with normal hematopoietic cells in the bone marrow. Alternatively FISH can be performed on cells that have been selected by either flow-cytometric cell sorting or immunomagnetic bead separation [23, 24, 27].
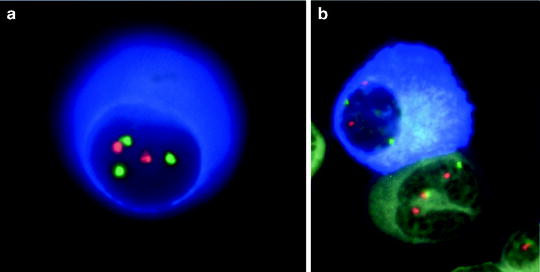
Fig. 30.1
FISH analysis of an MM cell. To be able to perform FISH on MM clinical samples it is necessary to select cells prior to FISH (e.g., using sorting or CD138+ magnetic beads) or to conjugate FISH with another way of limiting the scoring of cells to only plasma cells. We have developed the technique of concurrent immunofluorescent detection of monoclonal plasma cells using an anti-light chain (clone specific) antibody capable of detecting the cytoplasmic light chain. PC can be readily identified by the blue cytoplasm and FISH scoring is restricted to these cells. We call this technique cIg-FISH and can detect chromosome abnormalities restricted to the plasma cells. This allows detection of chromosome abnormalities in cases with a low level of plasmacytosis (e.g., in those with less than 1 % PC). It also allows for detection of chromosome abnormalities in non-PC [22] (Reprinted with permission of Informa Healthcare from Fonseca et al. [22])
Primary Genetic Aberrations
NH-MM and IgH Translocations
An increasing body of evidence suggests that chromosomal translocations at the immunoglobulin heavy chain locus (IgH) at 14q32 are of major significance in the pathogenesis of MM and MGUS [13, 21, 28–33] (Table 30.2). We and others have shown that these translocations are nearly universal in PC lines and are common in MM patient samples. Indeed, using interphase FISH, Nishida first reported that 14q32 translocations are found in a large fraction of MM patients [29]. Avet-Loiseau found translocations involving 14q32 in 60 % of patients [35], and we have documented IgH translocations in a similar proportion of patients [21, 36]. Others and we have also shown that translocations are also present in MGUS, often in cases without progression after many years, indicating that they are likely an initiating oncogenic event that precedes progression to frankly malignant MM [37, 38].
Table 30.2
International molecular classification of MM
Percentage of patients (%) | Clinical and laboratory features | |
|---|---|---|
Hyperdiploid | 45 | More favorable, IgG kappa, older patients |
Non-hyperdiploid | 40 | Aggressive, IgA lambda, younger individuals |
Cyclin D translocation t(11;14)(q13;q32) t(6;14q)(p21;32) t(12;14)(p13;q32) | 18 16 2 <1 | Upregulation of CCND1; favorable prognosis; bone lesions. Two subtypes by GEP Probably same as CCND1 Rare |
MMSET translocation | 15 | |
t(4;14)(p16;q32) | 15 | Upregulation of MMSET; upregulation of FGFR3 in 75 % unfavorable prognosis; bone lesions less frequent |
MAF translocation | 8 | Aggressive |
t(14;16)(q32;q23) | 5 | Confirmed as aggressive by at least two series |
t(14;20)(q32;q11) | 2 | One series shows more aggressive disease |
t(8;14)(q24;q32) | 1 | Unknown effect on outcome but presumed aggressive |
Unlcassified (other) | 15 | Various subtypes and some with overlap |
IgH translocations in MM can be classified as the common and uncommon (also called primary versus secondary or recurrent versus non-recurrent) and involve a number of non-random partner chromosomes [13, 32, 33, 39]. These translocations are thought to occur mostly in the IgH switch. In MM as in other B-cell neoplasias these translocations result in the juxtaposition of oncogenes with the powerful enhancer regions of 14q32, mostly the 3′ enhancer [28]. Most IgH translocations in MM are thought to be mostly balanced [28].
The diversity of the non-random chromosomal partners involved in these translocations is not well understood but likely reflects some combination of the presence of oncogenes near the translocation breakpoint and the inherent chromosomal structure surrounding the translocation breakpoints of the involved genomic loci. For instance, it seems likely that the 16q23 breakpoints result from genomic instability of the fragile site FRA16D [13, 40]. Generally, breakpoints in the partner chromosomes are widely scattered, frequently over several hundred large genomic regions [13]. Importantly, for all translocations that have been fully characterized, there is selective dysregulation and overexpression of one or more oncogenes that can be located up to 1 Mb from the translocation breakpoint. It is thought that the dysregulation is mediated by juxtaposition of the oncogene to one or more of the strong IgH enhancers, Eμ, 3′ Εα1, 3′ Εα2.
t(11;14)(q13;q32)
The t(11;14)(q13;q32) was the first well-recognized recurrent translocation in MM [1, 3]. This translocation is present in approximately 25 % of human MM cell lines [32] and dysregulates cyclin D1 and in some cases also a novel putative oncogene myeov [41]. Breakpoints in 11q13 are widely scattered and located as far as 330-kb centromeric to cyclin D1 on 11q13 [42]. Similar mechanistic upregulation of cyclin D1 has also been observed through the switch recombination-mediated excision and insertion of IgH constant and enhancer sequences at 11q13 as is seen in the cell line U266 [43]. Cyclin D1 is overexpressed in a fraction of myeloma patients as well, but immunohistochemical methods seem less able to detect the abnormality than cIg-FISH [44–46].
When interphase FISH techniques are used the incidence of t(11;14)(q13;q32) is 15–20 % [35, 47, 48]. While this translocation was initially thought to be associated with an adverse outcome, subsequent studies showed a trend toward a more favorable outcome for these patients [7, 30, 49, 50]. This translocation is also seen in the MGUS stage of the disease [38]. It is notable that there may be two major subtypes of t(11;14)(q13;q32) when these cases are studied via gene expression profiling (GEP), the so-called CD1 and CD2 groups [51]. Other variants of cyclin D gene translocations have also been described in MM, but their prognostic significance is undetermined [52].
The t(11;14)(q13;q32) is likely to be the most common IgH translocation in PC neoplasias; it is seen in 50 % of light chain amyloidosis cases, IgM MM, and primary plasma cell leukemia [53–55]. This has led to a current hypothesis that overall t(11;14)(q13;q32) is associated with a more favorable outcome, but under special circumstances and with the acquisition of additional genetic changes PC with t(11;14)(q13;q32) may exhibit more aggressive behavior [51].
The t(4;14)(p16.3;q32)
The t(4;14)(p16.3;q32) is also seen in approximately 25 % of human MM cell lines and in 15 % of human MM cases [14, 33]. This translocation has also been observed in MGUS albeit at a lower frequency than in MM [14, 33]. It results in the upregulation of the fibroblast growth factor receptor-3 (FGFR-3), which is located on der(14) [14, 33, 56]. In addition, hybrid mRNA transcripts including IgH sequences and the novel gene MMSET (WHSC1), which is homologous to the MLL gene that is involved in the pathogenesis of leukemia, are produced from der(4) [14, 33, 56]. The t(4;14) translocation provides the first example of a translocation that simultaneously dysregulates two putative oncogenes. The Ig/MMSET transcripts are readily detectable by RT-PCR and provide a useful tool for the detection of the abnormality [14, 33, 56]. The translocation breakpoints at 4p16.3 are dispersed over 200 kb and located centromeric to the FGFR3 gene and within the 5′ introns of MMSET [14, 33]. There is a selective expression of one FGFR3 allele in all informative cases, consistent with dysregulated expression of FGFR3 by the translocation [14, 33, 56]. Kinase activating mutations of FGFR3, identical to those causing thanatophoric dwarfism, have also been observed in the FGFR3 translocated allele [14, 33, 56].
This translocation is associated with more aggressive form of MM. Patients who harbor this translocation have a shorter duration of remission when treated with conventional forms of chemotherapy [34, 57]. In patients undergoing autologous stem cell transplant the translocation is associated with a very high rate of disease recurrence with most patients experiencing relapse less than a year after completing therapy [34, 57, 58]. Recent data suggest that some of the novel treatment agents for MM may overcome the negative prognostic implications of the translocation [59, 60]. Despite this excitement it appears that these observations resulted because of limited duration of follow-up of small groups of patients [59]. Although the novel agents such as bortezomib and lenalidomide can help a much larger group of patients, many of the old prognostic variables can still discriminate outcome, even when overall patients seem to do better [61–63] (Table 30.3).
Table 30.3
Prognostic outcomes of patients treated with bortezomib and standard genetic markers
Study | N | Outcome measured (p value) | Method and comments |
|---|---|---|---|
New diagnosis | |||
Vista [59] | 25/142 | Same CR 28 % | FISH detected HR-MM using t(4;14)(p16;q32), t(14;16)(q32;q23) and -17. Few events and needs long term follow up (HR 1.297) |
EFS 19.8 vs. 23.1 (0.5) | |||
Shaughnessy TT2 [64] | 35/341 | Hazard ratio of 2.34 (<0.001) | High risk determined by GEP. Largest series |
Shaughnessy TT3 [64] | 42/419 | Hazard ratio of 1.45 (1.74) | High risk determined by GEP. Largest series |
Avet-Loiseaua | 507 | Patients treated in IFM 2005-01 Arm B with VD induction. The t(4;14) still segregates outcome although better than VAD. Outcomes are not better with VD versus VAD for -17p patients. Translocation (p = 0.002) | |
t(4;14) in 106 (20.9 %) | t(4;14) PFS 28 vs. 35 (p < 0.02) | ||
3 year OS 73 % vs. 89 % (p = 0.002) | |||
-17 in 60 (11.8 %) | No difference with VAD | ||
Relapsed RR | |||
Jagannath (SUMMIT) [65] | 26/147 | EFS 24 vs. 33 (NS)a | High risk defined by karyotype chromosome 13. Low power as limited number of cases |
EFS 24 vs. 38 (0.36)** | |||
Jagannath (APEX) [65] | 11/74 | EFS 20 vs. 38 (0.2)a | High risk defined by karyotype chromosome 13. Low power as limited number of cases |
EFS 25 vs. 35 (NS)** | |||
Chang [66] | 65 | ORR 67 vs. 55 % (NS) | FISH detected HR-MM as in VISTA. Limited power |
Chang [67] | 85 | No difference in OS or PFS | Determined by FISH t(4;14), -17 and others. Very few cases with abnormalities studied |
Pineda-Roman [68] | 85 | t(4;14) class was favorable | Limited dataset and few cases with t(4;14). Category detected by GEP |
The t(14;16)(q32;q23) and Other MAF Variant Translocations
The t(14;16)(q32;q23) is present in approximately 5 % of human MM cases, has been reported in MGUS, and is found in 20 % of human MM cell lines and in PC leukemia [13, 36, 51]. The gene c-MAF has been shown to have transforming activity in chicken fibroblasts, and is dysregulated by this translocation [72]. Translocations at 16q23 also include variant translocations involving the lambda chain locus (8226 and XG-6) in which case the breakpoints are telomeric to c-MAF. Several factors support the notion that c-MAF is the important oncogene dysregulated by this translocation; upregulation of c-MAF is only seen in the cell lines with the t(14;16)(q32;q23), informative cells lines (KMS11 and RPMI8266) only express one allele, and c-MAF is bracketed by the 16q32 breakpoints of both the IgH and Ig lambda translocations [72]. Other translocations that involve other MAF genes have been described in a small proportion of patients [73, 74]. One study of the MAF-b translocated cases shows they also have a more aggressive phenotype [73].
The clinical significance of this translocation is similar to that of the t(4;14)(p16;q32). Even when seen in a small fraction of cases patients with t(14;16)(q32;q23) have a shorter survival and more aggressive clinico-pathologic features. The prognostic significance of t(14;16)(q32;q23) in the context of novel treatment agents is unknown since studies addressing its role have been so far inconclusive or limited.
Igλ Translocations in MM
Translocations involving the Igλ loci have been described in only a small fraction of MM tumors. For example, the SKY analysis of 50 advanced MM tumors by Sawyer et al. identified about 10 (20 %) of tumors with a translocation near the Igλ locus at 22q11 and perhaps 1 % or 2 % (<4 %) of tumors with a translocation near the Igκ locus at 2p11 [12].
Aneuploidy and Hyperdiploidy
MM is a disorder characterized by the frequent occurrence of aneuploidy (Fig. 30.2) [1–5, 23, 25, 76]. When taken as a whole aneuploidy is the most common chromosomal abnormality in MM [1–5, 23, 25, 27, 76]. Aneuploidy involves almost all chromosomes in the way of whole chromosome changes (trisomy), but sometimes even extra copies are gained also [1–5, 23, 25, 27, 76]. Although all chromosomes have been reported as trisomic in MM, only some show a high prevalence of trisomies and include all odd numbered chromosomes, with the exception of chromosome 13 [1–3, 20, 77, 78]. These trisomies can involve a variable number of clonal cells. The presence of several trisomies is what characterizes H-MM, where IgH translocations are less common [20, 21]. This hyperdiploidy state has been described in MGUS as well, and is believed to be one of the primary pathogenetic pathways of the disease, by yet to be determined molecular defects [79, 80]. It is clear that the specificity for these chromosome trisomies is not random and must confer certain advantage to cells that gain such copies [19–21, 79, 80]. The presence of these translocations also results in increased expression of genes located in these chromosomes [81].
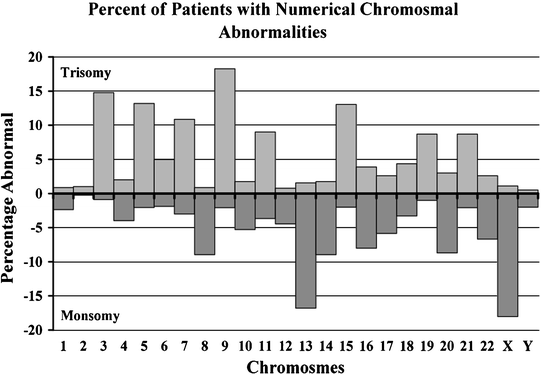
Overall hyperdiploidy is associated with a more favorable outcome [19, 82, 83]. Recently some secondary classifiers for prognosis of H-MM have been proposed including molecular cytogenetic and molecular signatures [82, 83]. In one study we were able to show that for H-MM the presence of secondary IgH translocations or deletions of 17p13 identify patients with higher risk disease [82]. Likewise we also did a GEP study that showed that patients with H-MM can be divided, in a validated fashion, into four groups, each with distinct prognosis implications [83].
Progression Events
Deletions of 13q
The presence of deletions of chromosome 13 has been associated with a poor prognosis in MM patients [6, 7]. The important sites for deletion at 13q14 have not been established and there is no proven pathogenetic role for 13q14 deletion in MM [77, 78]. In a report by the University of Arkansas, the presence of 13q abnormalities, as detected by CC analysis, was the single most important prognostic indicator [6, 7]. The net prognostic effect of chromosome 13 aberrations is much greater when these aberrations are detected via metaphases analysis as opposed to when FISH alone detects them [78, 84–86].
In most cases these deletions are monosomy or large deletions of the q-arm [77, 84, 87]. Other studies using aCGH as a basis to characterize these aberrations confirmed that most chromosome 13 deletions are monosomy with only a small fraction representing interstitial deletion [87]. It should be noted that while the prognostic power of chromosome 13 deletions detected by karyotype is much greater than when these are detected by FISH, FISH detection of chromosome aberrations is still prognostic. Nevertheless, FISH detection of chromosome abnormalities per se should not be considered an indicator of high-risk disease (see text to come) [88].
Deletions of 17p13.1
Deletions of the long arm of chromosome 17 at 17p13.1, with resulting loss of the tumor suppressor gene p53, are observed in about 10 % of MM cases and associated with a poor prognosis in multiple studies [36, 57, 61, 62, 89]. A striking correlation between p53 deletions and advanced and aggressive clinical features has been observed [36, 57, 61, 62, 89]. Current reports estimate a prevalence of p53 deletions in approximately 10 % of patients [36, 57, 61, 62, 89].
It is notable that the vast majority of patients with p53 deletions do not have overt evidence of coexistent mutation of the other allele [90, 91]. Mutations are rare at the time of diagnosis (2–3 %) but increase in prevalence with advancing stages of the disease [90–95]. However, recent data using high-density sequencing has revealed that up to 10 % of MM patients also have p53 mutations, raising the possibility that mutant p53 clones exist in a larger fraction of p53 deleted patients than currently suspected (Chapman, Personal Communication, June 2010).
Chromosome 1 Aberrations
Aberrations of chromosome 1 have been identified as important in determining outcomes of patients with MM. These include both gains of chromosome 1q regions as well as deletions of 1p [51, 96–99]. These abnormalities appear to increase in prevalence and importance as the disease progresses [51, 96–106]. Several regions have been identified as potentially containing genes of interest that may accelerate proliferation of cells, including CKS1B in 1q21 gains [51, 96] and deletion of p18 in the 1p region [98]. These aberrations have been proposed as important in the progression from MGUS to MM, although their precise role is still being debated [100–106].
NFKB Aberrations
Two separate efforts simultaneously identified abnormalities of the NF-kB pathway as important progression events for MM [107, 108]. The initial observations focused on the genetic inactivation of a negative regulator of the non-canonical NF-kB pathway, TRAF3 [107, 108]. This subsequently led to the identification of other genetic aberrations, all of which result in increased activation of the pathway [107, 108]. While the number of patients having such abnormalities is not precisely known, it is estimated that up to one half of patients have them [107, 108]. Keats hypothesized that patients harboring such aberrations would be more likely to be responsive to treatment with bortezomib [108]. While still in need of validation, patients with low level of expression of TRAF3 have a heightened sensitivity to bortezomib.
MYC Abnormalities
MYC is an oncogene that has been associated with development of plasmacytomas in murine models of the disease [109, 110]. Because of this MYC was initially believed to be a key element of the progression of the disease. Initial studies in MM revealed a low prevalence of MYC structural aberrations [12, 111], and these findings diminished enthusiasm in the pursuit of MYC as an important constitutive genetic aberration. Avet-Loiseau and colleagues, using a comprehensive FISH panel, identified a low frequency of MYC abnormalities with only 15 % of cases harboring such aberrations and without any important prognostic implications [111].
However other studies have pointed out MYC as a possible progression event for the disease [112–116]. Several studies from the Kuehl laboratory have shown a higher prevalence of MYC aberrations than suspected when more comprehensive genetic screening is used [112–116]. In particular MM cases with abnormal metaphases were shown to have a high prevalence of genetic aberrations and these were even more common in the human MM cell lines [112–116]. Still little information was available regarding the role of MYC in early stages of the disease [112–116]. Recently Chesi and Bergsagel developed a mouse model of MM that is based on post-germinal center activation of MYC (by releasing a pre-MYC stop codon via mutation) [117]. This model faithfully resembles the human disease in its clinico-pathologic features and responsiveness to drug therapy [117]. Using this model they identified an MYC signature that is also present in MM cases and is clearly more prevalent in cases of MM as opposed to MGUS [117]. While still unconfirmed MYC appears to be important as a progression event in early stages of the disease.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


