Keywords
Gene therapy, hematopoiesis, cytidine deaminase, doxycycline, miRNA, Ara-C
Background
Transfer of Drug-Resistance Genes into Hematopoietic Stem Cells
Given their ability to self-renewal, proliferate extensively, and differentiate into all the different mature cells of the lymphohematopoietic system in combination with their easy accessibility and a well-established technology for storage and transplantation, hematopoietic stem cells (HSCs) have been considered a particularly promising target population since the beginning of gene therapeutic activities in the mid-1980s. So far, HSC gene therapy has been successfully applied particularly to congenital diseases with manifestation in the hematopoietic system, such as severe X-linked combined immunodeficiency , adenosine deaminase deficiency , chronic granulomatous disease , and, recently, Wiskott–Aldrich syndrome and X-linked adrenoleukodystrophy .
Another attractive strategy to put HSC gene therapy to clinical use is the protection of the lymphohematopoietic system from the side effects of anticancer chemotherapeutic drugs by (over)expression of drug-resistance (CTX-R) genes. A number of such CTX-R genes have been identified, and for several of them, the potential to protect the lymphohematopoietic system from the associated cytotoxic agents has been firmly established in murine as well as large animal models . This includes mutant forms of dihydrofolate reductase (mutDHFR), the multidrug-resistance (MDR)-1 gene coding for the cellular efflux pump P-glycoprotein, and the gene coding for the DNA repair protein O 6 -methylguanine methyltransferase (MGMT), all of which have their specific advantages and disadvantages.
Mutant DHFR has been investigated for myeloprotective strategies since the late 1980s , and significant protection has been shown in vitro as well as in animal models . In the murine model, even in vivo selection of mutDHFR-transduced cells has been achieved . However, mutDHFR-induced myeloprotection is restricted to the small group of antifolate-type cytotoxic drugs such as methotrexate or trimetrexate. Although these agents induce considerable lymphotoxicity and are routinely administered for immunosuppression in a large number of disease states , generalized myelotoxicity is rarely encountered as the dose-limiting toxicity of folate antagonists, thus questioning the clinical relevance of mutDHFR for myeloprotection when used on its own.
On the other hand, MDR1 confers resistance against a wide variety of clinically highly relevant chemotherapeutic agents, such as anthracyclins, epipodophyllotoxins, taxoids, and vinca alkaloids, of which at least the first three groups are associated with profound and frequently dose-limiting myelosuppression. Effective protection from the toxicity of several of these agents upon MDR1 overexpression has been demonstrated for murine as well as human hematopoietic cells in vitro and in animal models ; however, effective transgenic MDR1 expression in clinical studies has been problematic .
Currently, the most promising CTX-R genes for myeloprotective purposes are MGMT mutants. Mutant MGMT gene transfer has proven highly efficacious for myeloprotection as well as in vivo selection in murine and several large animal models . Furthermore, a recent clinical trial demonstrated efficient myeloprotection and in vivo enrichment of genetically modified cells following mutant MGMT gene therapy in a cohort of glioblastoma patients, demonstrating extended survival in individual patients . However, the clinical indications for the associated drugs temozolomide and chloroethylnitrosoureas are rather limited, and apart from brain tumors, these drugs are only considered standard therapy for malignant melanomas . In addition, both drugs suffer from substantial non-hematopoietic toxicities and a considerable mutagenic potential .
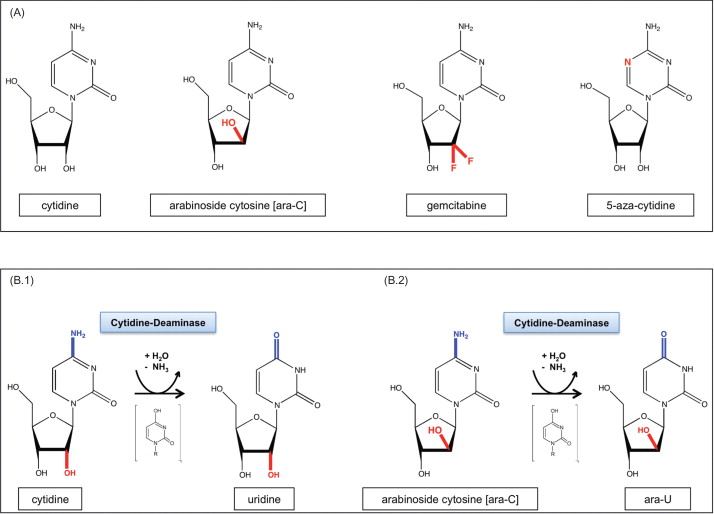
Cytidine deaminase (CDD) represents another interesting CTX-R gene. CDD protects cells from the clinically highly relevant cytotoxic deoxycytidine analogs cytosine arabinoside (1-β- d -arabinofuranosylcytosine (ara-C)), gemcitabine (2′,2′-difluorodeoxycytidine), and 5-azacytidine (5-aza-2′-deoxycytidine) ( Figure 29.1A ). In particular, ara-C has been utilized clinically for extended time periods and has been established as the most effective single agent in the treatment of acute leukemias. Importantly, ara-C has a predominantly hematopoietic toxicity profile, and when given at low to intermediate doses, hematotoxicity is the only side effect described. This toxicity profile may be related to the low CDD expression levels observed in hematopoietic progenitor/stem cells , whereas mature myeloid cells, and particularly granulocytes, represent the cells with the highest endogenous CDD expression within the hematopoietic system . A protective effect of CDD gene transfer in the context of ara-C and gemcitabine application has been established in murine and human clonogenic progenitor cells as well as in a murine in vivo CDD gene transfer model .
Nucleotide Metabolism and Nucleoside Analogs as Anticancer Drugs
Among the plethora of molecules required to maintain the complex structures and processes of living cells, nucleotides are of fundamental importance. All nucleotides share a common structure and consist of a nucleoside linked by a phosphoester to a phosphate group. The nucleosides in turn are composed of a pentose sugar and an organic nitrogen-containing base (nucleobase), with the purine bases adenine (A) and guanine (G) and the pyrimidine bases cytosine (C), thymidine (T), and uracil (U) constituting the most important ones.
Within a cell, nucleotides, in addition to their crucial role as components of polymeric nucleic acids such as DNA and RNA, exert a whole array of important functions. Thus, triphosphorylated forms of adenosine (ATP) and guanosine (GTP) serve as the most important sources of intracellular energy and regulate a large number of enzymatic reactions. Nucleotides also play important roles in cellular signaling pathways. For example, GTP activates guanine nucleotide-binding proteins (G proteins) as key mediators of signaling cascades, and cyclic nucleotides represent important secondary messenger molecules. In addition, nucleotides serve as integral structural elements of coenzymes (e.g., nicotinamide adenine dinucleotide), thereby controlling metabolic processes such as redox reactions .
In general, there are two pathways for an eukaryotic cell to replenish its nucleotide pool: de novo synthesis and nucleotide salvage (for schematic overview, see Figures 29.2 and 29.3 ). Whereas de novo biosynthesis starts from simple components (e.g., amino acids, H 2 O, pentose sugars, and phosphate) and results in the formation of newly synthesized nucleotides, in the salvage pathway preformed nucleobases or nucleosides, usually recovered from RNA or DNA degradation, are reutilized and converted to the nucleotide state. Uptake of salvage (deoxy)nucleosides may occur by two different transporter systems: a high-affinity transporter mediating active, sodium-dependent transport (concentrative nucleoside transporter) and a low-affinity transporter acting as a facilitated diffusion system (equilibrative nucleoside transporter) that are inhomogeneously distributed among different tissues. In mammals, de novo biosynthesis clearly represents the main source of nucleotides, whereas the salvage pathway becomes relevant, for example, in situations of high cell turnover, such as lymphoid differentiation .
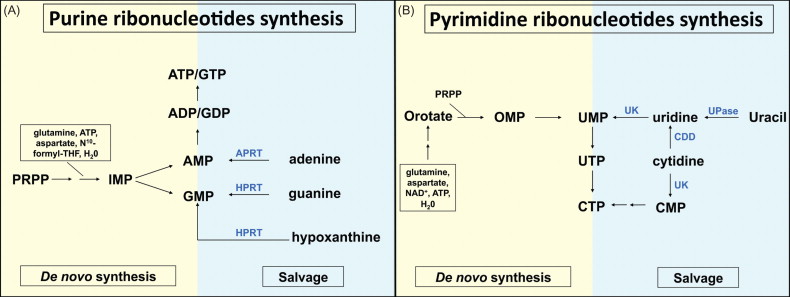
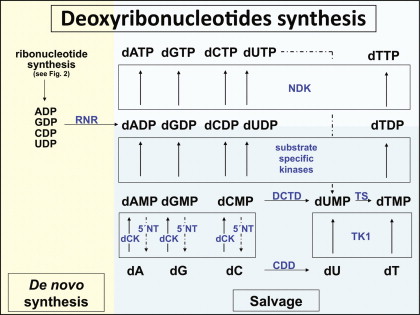
Nucleotides also can be degraded intracellularly either to uric acid (purine degradation) or β-alanine and β-aminoisobutyrate (pyrimidine degradation), and nucleotide and deoxynucleotide formation as well as breakdown is regulated through a complex network of feedback inhibition mechanisms, with nucleosides or nucleotides acting on multiple enzymes within the different pathways. Here, an example is the nucleotide-specific inhibition of ribonucleotide reductase ensuring the equal production of the different deoxynucleotides required for DNA synthesis ( Figure 29.3 ).
Given the universal importance of nucleotides for cellular function and survival, it is not surprising that nucleosides and their metabolites have been extensively evaluated as targets of anticancer drugs, while nucleoside analogs have replaced thymidylate synthetase inhibitors and antifolates as the most relevant group of antimetabolites in clinical cancer therapy. As a rule, nucleoside analogs represent prodrugs that, after entry into the cell and phosphorylation to the triphosphate state by the deoxynucleotide salvage pathway, exert their specific cytotoxic activities during or after incorporation into the DNA, where they interfere with DNA strand elongation, replication, or repair processes . Whereas the therapeutic activity of the traditional nucleoside analogs ara-C and 5-mercaptopurin has been largely restricted to acute leukemias, newer drugs such as fludarabine, cladribine, and particularly gemcitabine have a much broader spectrum of antitumor activities. Also, specific resistance factors to nucleoside analog therapy have been identified, and several of them represent bona fide CTX-R genes suitable for myeloprotective gene therapy approaches ( Figure 29.4B ).
Cytidine Deaminase: Structure, Function, and Role in Therapy with Cytidine Analog
CDD (EC 3.5.4.5.) is an enzyme involved in the nucleotide salvage pathway and catalyzes the deamination of cytidine and deoxycytidine to uridine and deoxyuridine, respectively ( Figure 29.1B.1 ). The CDD gene is located on the small arm of chromosome 1 (1p35–p36.2) and extends over approximately 31 kb . The gene consists of four exons with lengths of 60–450 bp that are separated by three introns of 4.6–16 kb . The first exon includes the 5′ untranslated region (5′ UTR) and parts of the coding region, whereas the fourth exon encodes for the C-terminus of the protein and the 3′ UTR of the respective mRNA . The CDD gene encodes a polypeptide of 146 amino acids with a molecular mass of approximately 16 kDa. Alignment of the CDD protein from different species demonstrates a high degree of conservation, especially for the catalytic domain down to the prokaryocyte state. The catalytic activity of the enzyme requires zinc, and studies reveal that functional CDD acts as a tetramer of four identical subunits .
CDD exerts its enzymatic activity not only on its physiologic substrates cytidine and deoxycytidine but also on a number of cytidine analog-type drugs, such as ara-C ( Figure 29.1B.2 ), gemcitabine, and 5-azacytidine, which may be inactivated by the enzyme . In this context, as early as 1971, Steuart and colleagues described high levels of CDD in the leukemic blasts of acute myeloid leukemia (AML) patients, relapsing after ara-C treatment , thus introducing the notion of CDD as a resistance factor to ara-C activity in leukemia therapy ( Figure 29.4A ). Although the correlation between CDD activity in AML blasts and clinical response to ara-C therapy has been confirmed by several groups and particularly pretreatment CDD activity has been described to predict therapeutic outcome , other groups have failed to establish this correlation . In several studies, this may have been due to the insufficient numbers of individual patients investigated, whereas other studies have demonstrated a correlation of low CDD activity levels caused by genetic CDD variants with therapy-associated side effects . Thus, a higher treatment-related mortality and potential ara-C dose adjustments counteracting the positive effect on ara-C activation have to be considered when assessing the overall effect of low CDD activity levels. Clinically relevant CDD-associated drug resistance has also been demonstrated for other cytidine analogs and other diseases. Thus, in myelodysplastic syndrome patients resistant to 5-aza-deoxycytidine (decitabine), a higher CDD/deoxycytidine kinase (dCK) ratio in comparison to that of decitabine responders was demonstrated , and in patients with non-small-cell lung cancer (NSCLC) the therapeutic response to gemcitabine correlates with genetic variants of the CDD gene .
The previously discussed data highlight the relevance of CDD polymorphisms and their influence on CDD enzymatic activity in the context of ara-C application. Approximately 20 different single nucleotide polymorphisms (SNPs) have been identified within the Caucasian, African-American, Japanese, and Korean populations . A79C/Lys27Gln or T435C and the promoter SNPs A-92G and C-451T represent the most common SNPs, with frequencies of up to 60% in a cohort of 360 AML patients . Although the majority of these SNPs in functional studies showed similar enzyme activity as the wild-type CDD alloenzyme, for the A79C/Lys27Gln and several of the promoter SNPs, a reduced capacity to deaminate ara-C and gemcitabine was demonstrated along with a lower affinity of the variant Gln27 allozyme to its substrate . In AML, CDD polymorphisms associated with reduced enzyme activity such as A79C/Lys27Gln or the C-451T/promoter SNP have been associated with superior therapeutic outcome as well as increased therapy-related side effects , although this has not been confirmed universally . Thus, for gene therapy approaches, CDD polymorphisms clearly bear some relevance because they may influence enzyme activity and thereby alter protection capacity . However, to date, no variants with increased enzyme activity have been described, and most studies aiming for the transgenic overexpression of CDD have been performed with the wild-type cDNA.
Gene Transfer of Cytidine Deaminase into the Hematopoietic System
Efficacy of Cytidine Deaminase Gene Transfer In Vitro and in Animal Models
Early Cell Line and Murine In Vitro Studies
The first studies describing a protective effect of CDD (over)expression on cytidine analog-induced cytotoxicity used murine fibroblast cell lines transfected with the cDNA of human (h)CDD and were reported in 1996 . Although these studies varied considerable with regard to the degree of CDD overactivity achieved in individual CDD expression clones (50- and 2.5-fold, respectively) and correspondingly also in the degree of ara-C resistance obtained (100- and 3.0-fold, respectively), they clearly established a gene dose-dependent cytoprotective effect of transgenic CDD overexpression. No obvious transgene-related toxicity was detected in CDD-transfected fibroblasts when morphology and growth characteristics were compared to those of control cells. These observations were confirmed in the same year by two studies employing Moloney murine leukemia virus (MMLV)-based γ−retroviral gene transfer technology to express hCDD in 3T3 fibroblasts, hematopoietic CCRF-CEM cells, as well as primary murine bone marrow cells . Here, constitutive overexpression of hCDD from an LXSN vector backbone resulted in 2.1- and 4.5-fold increased ara-C resistance in CCRF-CEM and 3T3 cells, respectively, and CCRF-CEM cells were also protected from gemcitabine (2.4-fold). CDD as the functional basis for the observed ara-C resistance was confirmed by complete reversibility of the effect by the highly specific, competitive CDD inhibitor tetrahydrouridine . Furthermore, Momparler and colleagues described 10- and 1,000-fold increased ara-C resistance in 3T3 fibroblast and clonogenic primary bone marrow cells, respectively, utilizing an MFG-based construct . These results were confirmed and extended by a number of in vitro studies published during the following 5 years, all of which applied γ−retroviral gene transfer technology to transduce fibroblasts, hematopoietic cell lines, and primary murine hematopoietic cells . Although these studies recapitulated increased ara-C resistance upon CDD gene transfer and extended these data to other cytidine analog-type cytotoxic drugs, such as gemcitabine and 5-azacytidine , the levels of protection conferred by CDD overexpression varied considerably, ranging from 2.5- to 20-fold in hematopoietic cell lines such as WEHI-3 and L1210 cells to 2- to 10-fold in primary clonogenic cells . The rather low protection rates observed in some of these studies probably reflect the high endogenous CDD background level in WEHI-3 cells or suboptimal gene transfer efficacies for primary cells . CDD gene transfer was also investigated in a murine transplant model in which recipients of CDD-transduced bone marrow cells were subjected to ara-C treatment. However, despite successful gene transfer, only moderately increased ara-C resistance was detected in clonogenic progenitors harvested 1 year after transplantation. Furthermore, no convincing evidence for in vivo myeloprotection was observed, most likely due to insufficient long-term expression of the hCDD transgene .
CDD Overexpression in Human CD34 + Progenitor/Stem Cells
Because forced overexpression of CDD in human primitive hematopoietic cells represents a prerequisite for the clinical application of CDD gene transfer, the effect of CDD overexpression was also evaluated in human CD34 + stem/progenitor cells. Whereas efficient gene transfer into primitive hematopoietic stem cells traditionally has been much more difficult to achieve in the human compared to the murine system, many of the hurdles responsible for this discrepancy, such as low HSC availability, insufficient quality and purity of starting populations, or low transduction efficiency due to insufficient transduction protocols for human HSCs, have been overcome by meticulous studies in vitro as well as in murine, canine, or nonhuman primate models. Consequently, optimized transduction strategies currently apply optimized cytokine combinations , fibronectin (retronectin) co-culture , or vector particles pseudotyped with suitable envelope proteins . Thus, it took until 2005 to establish definite proof of the protection of primary human hematopoietic cells by CDD gene transfer. Transduction of umbilical cord- or peripheral blood-derived CD34 + stem/progenitor cells with an SFFV-based vector harboring the hCDD-cDNA resulted in pronounced hCDD overexpression and a significantly increased resistance of transduced progenitor cells to ara-C (20–100 nM) and gemcitabine (10 nM), as determined by the effect on clonogenic growth of progenitor cells differentiated along the erythroid as well as the myeloid lineage .
CDD Gene Transfer in Murine In Vivo Transplant Models
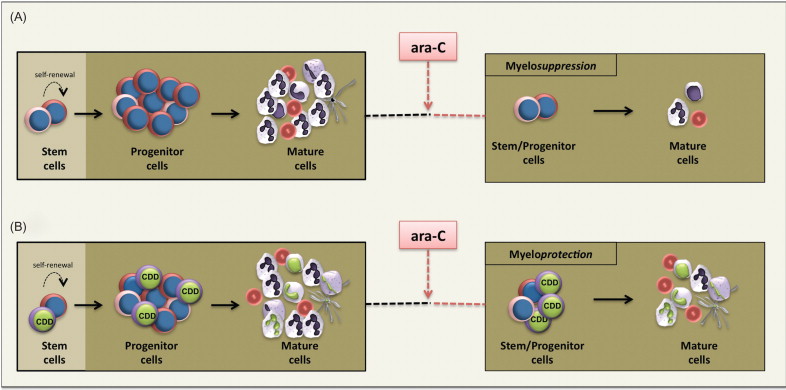
Clearly, in vitro protection and selection studies cannot always be considered predictive for the in vivo situation within an intact organism, with its complex network of finely tuned regulatory mechanisms and interactions involving different cell types and molecules. For the lymphohematopoietic system, HSC transplant models have convincingly filled this gap . Utilizing this technology, not only has long-term expression of CDD in the hematopoietic system been achieved but also constitutive overexpression of CDD in the murine lymphohematopoietic system was proven to confer efficient and significant protection against short-term high-dose (500 mg/kg, days 1–4, i.p.) as well as prolonged low-dose (30–60 mg/kg, days 1–10;,i.p.) ara-C application. For the short-term high-dose schedule, granulocyte nadirs of 2.9±0.6 versus 0.7±0.1/nl ( p <0.001) and thrombocyte nadirs of 509±147 versus 80±9/nl ( p =0.008) were observed for the CDD versus the control group, respectively , and similar differences (granulocytes: 2.5±0.5 vs. 0.2±0.1/nl, p =0.002; platelets: 546±76 vs. 112±35/nl, p =0.002; all CDD vs. control group) were observed following prolonged low-dose treatment . Moreover, animals overexpressing CDD in their hematopoietic system were also protected from otherwise lethal gemcitabine doses, and a 24- to 149-fold increased cytoplasmic CDD activity was observed in the bone marrow and spleen cells of primary recipients . Although these data reflect the excellent expression levels achievable with SFFV/MESV-based vectors in hematopoietic cells, these very high levels also may have contributed to the transgene-related toxicities observed in these studies. Interestingly, despite the profound myeloprotective effects observed in these studies, no long-term in vivo selection of CDD overexpressing cells was achieved .
Predictions from Preclinical Models for Clinical Application Scenarios
In general, in vitro studies employing cell lines or murine clonogenic progenitor growth have demonstrated hCDD-mediated ara-C resistance at concentrations of 10–500 nM . The only exception was an early study by the Momparler group reporting resistance to ara-C doses of up to 50 µM , but this group later described resistance only in the aforementioned dose range . CDD-mediated resistance to ara-C doses ranging from 60 to 500 nM was also observed for primary human clonogenic cells . These similarities are not surprising because for both human and murine progenitor/stem cells, low CDD activity has been demonstrated , and in both species the hematopoietic system represents a critical target organ for ara-C and gemcitabine toxicity . These data strongly suggest that in the clinical situation, protection of hematopoietic cells at least from conventional-dose ara-C should be possible by CDD overexpression. Ara-c therapy conventionally is applied at doses of 100–200 mg/m 2 given consecutively for 3–7 days, and when delivered as continuous infusion, this results in steady-state plasma concentrations of 100–1200 nM. Because the most reliable parameter to predict ara-C cytotoxicity (within certain limits) is the product of exposure time and drug concentration, this ara-C exposure was clearly exceeded in the experimental in vitro studies. In clonogenic assays, hematopoietic cells were protected from ara-C doses of up to 500 nM (and even up to 2000 nM in our recent studies utilizing advanced lentiviral vectors; unpublished data) applied for 10–14 days. The Rattmann study furthermore suggests that protection in the clinical situation should also be possible from high-dose ara-C application (3 g/m 2 i.v., six to eight doses at 12-hr intervals). This notion, however, should be addressed with caution because different application schedules (once daily i.p. in the murine model vs. twice daily i.v. in the clinical situation) have to be taken into account.
Combinations of CDD with other CTX-R Genes
Given the fact that modern anticancer chemotherapeutic regimens almost inevitably combine a number of cytotoxic drugs to reduce agent-specific toxicities and delay the emergence of therapy-resistant tumor cells, simultaneous (over)expression of drug-resistance genes appears to be a suitable strategy. With respect to CDD in particular, combinations with mutDHFR have been investigated utilizing MMLV-based retroviral backbones. Combining the mutDHFR- and CDD-cDNA via an internal ribosomal entry site or a thrombin cleavage site sequence, protection against methotrexate as well as ara-C has been demonstrated . Indeed, successful treatment of human lymphoma cells has been reported in a humanized mouse model utilizing CDD/mutDHFR cotransduced murine bone marrow cells to protect the hematopoietic system from the combined application of ara-C and methotrexate , although the exact individual contribution of the two resistance genes to the overall protective effect remained unclear in this study . Overexpression of hCDD has also been combined with expression of glutathione S -transferase A3 to protect from combined nitrogen mustard and ara-C treatment , although this concept never progressed beyond in vitro cell line studies. Anyway, given the successful application of mutMGMT gene transfer in a recent phase I study in glioblastoma patients , mutMGMT appears to be a much more promising combination partner to achieve myeloprotection from alkylating agents and in vivo selection of drug-resistant hematopoietic cells in the context of CDD gene transfer. This concept may be exploited in the treatment of malignancies responsive to ara-C as well as BCNU, such as salvage therapy of aggressive lymphomas based on the DexaBEAM (dexamethasone, BCNU, etoposide, ara-C, melphalan) regimen. Furthermore, considering the dominate role of ara-C in the therapy of AML and high-risk myelodysplastic syndromes, the fact that anthracyclins are the only other agents with a similar therapeutic efficacy in these diseases, and the profound and long-lasting myelotoxicity encountered with the combined use of these agents, clearly MDR1 appears to be another highly promising combination partner for CDD gene therapy. This strategy is currently being evaluated by our group in representative cell line and in vivo animal models. An overall overview of cytidine deaminase in myeloprotective gene therapy is given in Table 29.1 .
| Model | Cell Type | Gene Expression Systems | Years | References | |
|---|---|---|---|---|---|
| In Vitro | |||||
| Cell lines | 3T3 fibroblast | Retrovial | Constitutive | 1996, 1998 | |
| CCRF-CEM (hematopoietic) | Retrovial | Constitutive | 1996 | ||
| Various | Plasmid (cDNA) | Constitutive | 1996 | ||
| Fibroblasts | Plasmid (cDNA) | Constitutive | 1996 | ||
| NIH 3T3 fibroblast a | Retrovial | Constitutive | 1998–2000 | ||
| WEHI-3 (hematopoietic) | Retrovial | Constitutive | 1999 | ||
| L1210 (hematopoietic) | Retrovial | Constitutive | 2001 | ||
| Human lung carcinoma cells | Retrovial | Constitutive | 2002 | ||
| Bladder cancer cell line | Plasmid (cDNA) | Constitutive | 2003 | ||
| 32D (hematopoietic) | Lentiviral (third generation) | Inducible | 2012 | ||
| Primary mouse | Primary bone marrow cells (BMCs) | Retrovial | Constitutive | 1996–2001 | |
| Primary BMCs a | Retrovial | Constitutive | 1998–1999 | ||
| Bone marrow stromal cells | Retrovial | Constitutive | 2002 | ||
| Primary hematopoietic stem cells (HSCs) | Lentiviral (third generation) | Inducible | 2012 | ||
| Primary human | Cord blood and peripheral blood-derived progenitor cells | Retrovial | Constitutive | 2005 | |
| In Vivo | |||||
| Primary mouse | Primary hematopoietic BMCs | Retrovial | Constitutive | 1998 | |
| Primary BMCs a | Retrovial | Constitutive | 2004 | ||
| Primary HSCs | Retrovial | Constitutive | 2006, 2012 | ||
| Hematopoietic cell line 32D/primary HSCs | Lentiviral (third generation) | Inducible | 2012 | ||
Drawbacks Associated with Cytidine Deaminase Gene Transfer
Toxicities Related to the CDD Transgene
Although preclinical studies have clearly established the myeloprotective potential of CDD (over)expression, the problems associated with constitutive CDD overexpression in the lymphohematopoietic system have also been displayed. Using a conventional long terminal repeat (LTR)-driven γ-retroviral vector, Rattmann and colleagues observed a significant reduction of gene marked lymphocytes relative to myeloid cells in animals transplanted with CDD-overexpressing HSCs, which was not detected in a GFP control group. This notion of CDD-induced lymphotoxicity was further supported by significantly decreased peripheral blood lymphocyte counts in animals transplanted with CDD-modified HSCs transduced at high gene transfer rates. A potential biochemical mechanism for this lymphotoxicity has been suggested by recent studies in mice devoid of dCK activity . dCK acts as another enzyme of the nucleotide salvage pathway and represents the physiologic “counter-enzyme” of CDD ( Figure 29.3 ) because it catalyzes the phosphorylation of deoxycytidine to the monophosphate state and thereby competes with cytidine deaminase for this substrate. Thus, both CDD overexpression and reduced dCK activity directly result in decreased CMP levels. Whereas the deoxyribonucleotide salvage pathway is dispensable for most developmental processes including embryo- and organogenesis, it appears to play a critical role during early T and B cell development when T cell receptor or VDJ recombination is followed by massive cellular proliferation and clonal expansion . Mice deficient in dCK have a two- or threefold deficiency in both Hardy fractions A–D (B220 + IgM − ) and E–F (B220 + IgM + ) B cell precursor populations as well as an accumulation of thymocytes at the DN3 stage (CD4 − /CD8 − /CD44 low /CD25 + ), resulting in a reduction of the DN4 (CD4 − /CD8 − /CD44 − /CD25 − ) population and therefore a block in early T and B cell development .
In addition, a mild to moderate myelotoxicity of CDD overexpression has been described, most likely related to the negative feedback loop instituted by the release of high levels of CDD by mature human granulocytes, which in turn inhibits the differentiation and proliferation of granulocyte–macrophage colony-forming unit (CFU-GM) cells and thereby regulates late steps of myeloid differentiation . Whereas in the original report, the growth inhibition of murine and human CFU-GMs by exogenously supplied CDD required supraphysiologic concentrations of thymidine in the media , our group described moderate inhibition of human CFU-GM formation following transgenic CDD overexpression also at physiologic thymidine levels . Furthermore, we observed a mild reduction in granulocyte counts in our murine in vivo CDD gene transfer model, particularly in experiments showing high transduction rates , although in the in vivo situation, the inhibitory effect of CDD on myeloid progenitor and precursor cells may become ameliorated by counter-regulatory mechanisms of granulocyte homeostasis.
Lack of Efficient In Vivo Selection Capacity
Whereas HSC gene therapy has been applied successfully to congenital diseases of the hematopoietic system, in several of these disease entities the therapeutic transgene itself conferred an in vivo growth and survival advantage to gene-corrected HSC or early lymphoid progenitor populations . This may constitute a dilemma for diseases lacking a functional manifestation on the HSC or primitive progenitor level, such as Gaucher’s disease, mucoviscidosis, or hemoglobinopathies. Here, genetic correction per se will not provide a selective advantage to long-lived cells in vivo , and enrichment of genetically corrected hematopoietic cells by other means may be required. In this context, CTX-R genes such as mutant MGMT have been considered as powerful selectable markers allowing for an efficient selection of gene-modified cells. With regard to CDD, efficient in vitro selection of CDD-modified cells by ara-C, gemcitabine, or 5-azaycytidine exposure has been shown for various cell types, including murine L1210 leukemic cells and primary adherent bone marrow stromal cells, and in several of these studies an enrichment of transgene-positive cells of greater than 99% was demonstrated . More important, significant in vitro selection of CDD-transduced primary human hematopoietic progenitor cells was observed upon culture of the cells in medium containing 30–100 nM ara-C for 4 days . Despite these successful in vitro studies, in vivo enrichment of CDD-modified hematopoietic cells has been problematic. This became obvious from the studies of Rattmann and colleagues that were performed in a murine transplant model. Here, in sharp contrast to the marked myeloprotection, a complete lack of in vivo selection was observed with a short-term high-dose ara-C treatment schedule . This suggests that the protective effect exerted by CDD overexpression was achieved primarily on the level of progenitor or more mature cells rather than the stem cell compartment. Indeed, relatively moderate stem cell toxicity in comparison to that of alkylator-type drugs such as busulfan or BCNU has been described for ara-C —an observation readily explained by the relative quiescence of repopulating HSCs and the S-phase-specific activity of ara-C. Such a “stem cell-sparing” activity of ara-C is also suggested by its clinical toxicity profile. Here, following repetitive cycles of high-dose ara-C application (3 g/m 2 , six to eight doses), a profound and long-lasting myelosuppression almost inevitably is followed by a complete hematopoietic reconstitution, although more detailed studies in murine transplant models have demonstrated a 4-fold decrease in long-term repopulating capacity and a 10-fold reduction in pre-CFU activity following dose-intensive ara-C therapy . On the other hand, repetitive low- to intermediate-dose ara-C application has been reported to increase cycling and thus ara-C susceptibility of HSCs . This was recently investigated in a murine CDD gene transfer/HSC transplant model. Here, ara-C doses of 30–60 mg/kg i.p. given repetitively for 10–20 days resulted in significant in vivo selection with an up to 6-fold increase of CDD-transduced cells in the peripheral blood. However, this effect was only transient, suggesting selection on the level of early progenitors rather than HSCs . One approach suggested to promote stable and long-time in vivo selection of hematopoietic cells by the CDD/ara-C selection system is the priming of HSCs prior to selection chemotherapy to recruit these cells into the active cycle and thereby render them more susceptible to ara-C treatment. To this point, pretreatment with granulocyte colony-stimulating factor, interferon-α, or cytotoxic drugs such as 5-fluorouracil and cyclophosphamide may be explored . Another option would be ara-C application during the immediate post-transplantation myeloablative period. In this situation, maximal cycling of HSCs is encountered, and recently this has been exploited successfully to achieve robust in vivo HSC selection by the HPRT/6-thioguanine selection system .
Insertional Mutagenesis
Insertional mutagenesis has been described in several HSC gene therapy studies and clearly represents a major concern for all studies utilizing integrating vector systems. This carries a particular relevance for CDD gene transfer applications because in this setting, insertional mutagenesis may give rise to drug-resistant leukemic cells. In clinical studies, insertional mutagenesis has been observed specifically for γ-retroviral constructs expressing the therapeutic gene directly from the strong viral promoter/enhancer sequences situated in the U3 region of the LTR. In this context, insertion of the vector upstream of a cellular gene has been observed to lead to an upregulation of cellular (onco)genes such as lmo2 or evi1 mediated by the viral enhancer . Significant reduction of the mutagenic risk can be achieved by the use of improved γ-retro- or lentiviral vector design, such as self-inactivating constructs harboring inactivating deletions in their 3′ LTR U3 promoter/enhancer region , and the use of internal promoters derived from housekeeping genes . We are currently investigating such safety improved constructs in the context of CDD gene transfer. However, given the risk of generating CDD-overexpressing leukemic cells, incorporation of inducible suicide genes such as herpes simplex virus thymidine kinase (HSV-TK) or a genetically engineered variant of caspase 9 (i-caspase) into the gene transfer vectors should be considered as a fail-safe mechanism.
Inadvertent Transduction of Leukemic Cells
Because CDD-associated drug resistance is observed primarily for agents that exert their therapeutic activity in acute leukemias and other hematological malignancies, inadvertent transduction of leukemic cells present in the cell preparation used for the ex vivo genetic modification procedure constitutes another potential problem of CDD gene transfer. Thus, currently it appears preferable to use this strategy in combination with an allogeneic bone marrow transplant approach because it is state-of-the-art for high-risk acute myeloid or lymphoid leukemias and myelodysplasias . If CDD gene transfer is applied in an autologous setting, suicide genes may be considered as a fail-safe mechanism.
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Ethics in Translational Gene Transfer Research
Clinical Trials Using LV-P140K-MGMT for Gliomas
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


