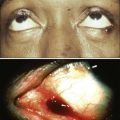
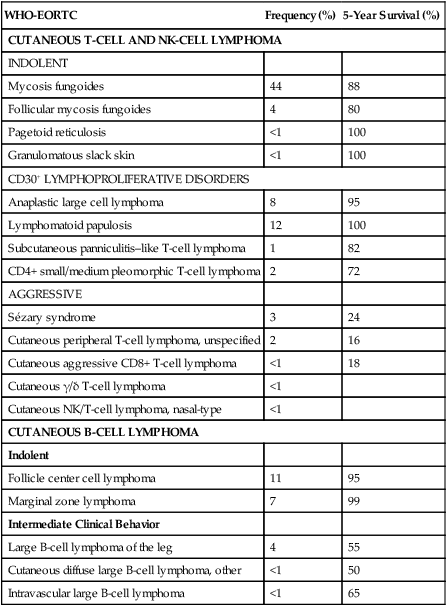
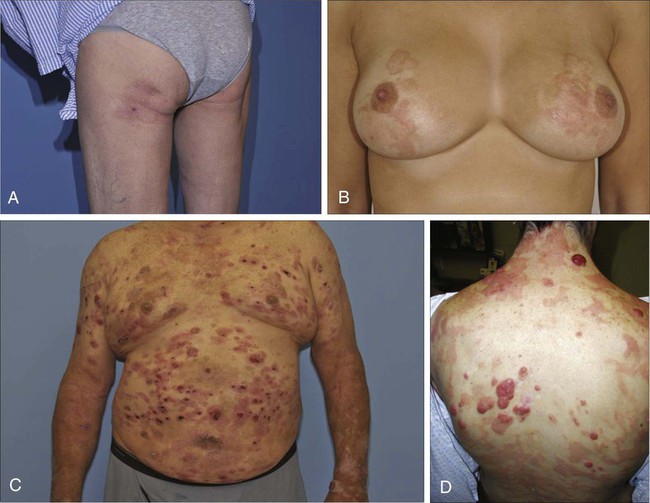
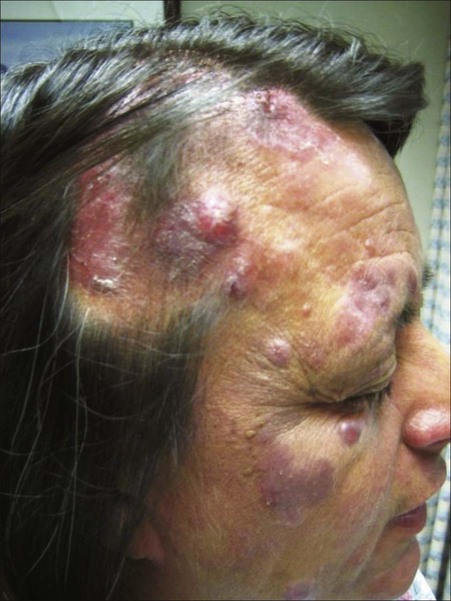
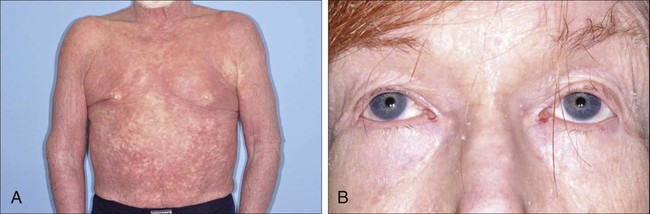
Christiane Querfeld and Steven T. Rosen • Cutaneous lymphomas represent 3.9% of non-Hodgkin lymphomas. • The annual incidence of cutaneous T-cell lymphoma (CTCL) in the United States is approximately 9.6 cases per 1 million population with a median age of 60 years at initial presentation. • CTCL accounts for up to 71% and cutaneous B-cell lymphoma (CBCL) accounts for up to 29% of all cutaneous lymphomas. • CTCL may be indolent as in mycosis fungoides (MF) or aggressive as in Sézary syndrome (SS). • The malignant T cells in MF/SS are characterized by a CD4+ T-helper (Th)/memory cell phenotype with frequent loss of CD7 and/or CD26 and the expression of skin homing markers such as CLA, CCR4, and CCR10. MF/SS demonstrate an altered immune biology and the accumulation of cytogenetic abnormalities during disease progression with a predominant Th2 cytokine profile in advanced stages. • Primary CBCLs such as cutaneous follicle center lymphoma have failed to show similar alterations compared with their nodal counterpart. The prognosis of both primary cutaneous follicle center lymphoma and cutaneous marginal zone lymphoma is usually excellent. • The gene expression profile of primary cutaneous large B-cell lymphoma, leg type, is similar to that of nodal diffuse large B-cell lymphoma (DLBCL) arising from germinal center or post–germinal center–activated B cells with constitutive nuclear factor–κB (NF-κB) pathway activation and strong expression of the IRF4/MUM1 transcription factor and carries a worse prognosis. • All patients with cutaneous lymphoma should initially be seen and co-managed by a multidisciplinary cutaneous lymphoma team consisting of members from dermatology and oncology with the support of other health care professionals such as radiation oncologists, pathologists, and clinical psychologists. • Routine evaluation should include complete physical examination, complete blood cell count with differential, and chemistry panel with lactate dehydrogenase (LDH) level, skin biopsy for histology, immunophenotyping and gene rearrangement studies, and lymph node biopsies in cases with enlarged nodes at presentation. • Imaging studies and flow cytometry panel of peripheral blood should be reserved for patients with clinical and laboratory findings suggestive of systemic disease and/or prominent lymphadenopathy. Bone marrow biopsy is a consideration in advanced-stage disease. Histopathological and molecular results should be correlated with clinical findings and patients classified according to the World Health Organization/European Organization of Research and Treatment of Cancer (WHO/EORTC) consensus classification. • Early-stage MF usually responds well to skin-directed therapies such as phototherapy, radiation, topical nitrogen mustard, retinoid, or corticosteroid skin creams. • Treatment for patients with advanced stages of MF or SS generally requires systemic therapies, including biological or immune therapies, histone deacetylase inhibitors, conventional chemotherapeutic agents, or combinations of these agents. A variety of non-Hodgkin lymphomas can involve the skin, either primarily or secondarily. Primary cutaneous lymphomas present in the skin with no evidence of extracutaneous disease at the time of diagnosis. They comprise a heterogeneous group of cutaneous T-cell lymphomas (CTCLs), cutaneous B-cell lymphomas (CBCLs), natural killer (NK) cell, and precursor hematopoietic neoplasms with distinct variability in clinical presentation, histopathology, immunophenotyping, gene rearrangement, and prognosis. The diversity of clinical and pathological manifestations among subsets of cutaneous lymphomas has led to much controversy over its diagnosis and classification and to the proposition of new variants and the establishment of consensus guidelines by a joint effort of the World Health Organization and European Organization for Research and Treatment of Cancer (WHO/EORTC) in 2005.1 These guidelines were incorporated into the revised fourth WHO classification of tumors of hematopoietic and lymphoid tissues in 2008 using the existing framework for nodal lymphomas; however, differences remain, particularly to the subclassification of CBCLs.2 Based on the WHO/EORTC classification, cutaneous lymphomas are classified as indolent, intermediate, or aggressive.1,3 The two most commonly recognized types of CTCL are mycosis fungoides (MF), which is generally indolent, and Sézary syndrome (SS), which is an aggressive and leukemic form of disease (Table 107-1). MF and SS together comprise 53% of all cutaneous lymphomas and are often synonymously named as CTCL. Notably, cutaneous manifestations of human T-cell lymphotrophic virus type I (HTLV-I)–associated adult T-cell lymphoma/leukemia (ATLL) occur in about 50% of cases and can mimic a slowly progressing course of MF.4,5 The second common CTCL type encompasses the spectrum of primary cutaneous CD30+ lymphoproliferative disorders, including lymphomatoid papulosis and cutaneous anaplastic large cell lymphoma; this CTCL type comprises 20% of all cutaneous lymphomas. Other, rarer types include the subcutaneous panniculitis–like T-cell lymphoma, which is generally indolent, and the group of primary cutaneous peripheral T-cell lymphomas (PTCLs) that includes the provisional entities cutaneous aggressive epidermotropic CD8+ T-cell lymphoma, cutaneous γ/δ T-cell lymphoma, and cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma, with the last generally recognized as more indolent than the others. Table 107-1 The WHO/EORTC Consensus Classification for Primary Cutaneous Lymphomas with Relative Frequency and 5-Year Survival Rate Modified from Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768–85. Cutaneous lymphomas represent about 3.9% of all non-Hodgkin lymphomas. Frequency and survival data used in the WHO/EORTC classification are based on Dutch and Austrian registries.1 Cutaneous lymphoma incidence patterns were studied using data from 3884 patients from 16 Surveillance Epidemiology and End Results (SEER) program registries in the United States during 1980 through 2005.6 Data revealed that CTCLs account for 71% and CBCLs account for 29% of all lymphomas. MF accounted for 38% of all CTCLs, followed by cutaneous PTCL (20%) and CD30+ lymphoproliferative disorders (10%). The age-adjusted annual incidence of CTCLs in the United States with most cases classified as MF has increased from 2.8 per million (1973-1977) to 9.6 per million (1998-2002) according to data from Criscione and Weinstock, and from 5.0 per million to 12.7 per million, according to Bradford and associates.6,7 MF is the most common type of CTCL, with a predominance of male patients of approximately 2 : 1 and a predominance of African-American patients of 1.6 : 1. The median age at presentation is between 50 and 70 years with only a small number of cases occurring in children and young adults. CBCL incidence rates are highest in non-Hispanic whites with a male predominance and an exponential increase with age. The most common primary CBCL subtypes are cutaneous DLBCL, follicle center cell lymphoma, and marginal zone lymphoma, comprising 40%, 30%, and 25% of all cases of CBCL, respectively. Despite rising incidence rates of CTCL, its etiology remains elusive. It has been suggested that the condition results from persistent antigen stimulation; however, its antigen-driving process is not known. Microbiological, environmental, occupational, and lifestyle factors have been suggested in the etiology of CTCL, but none has yet been verified.10–10 Unlike ATLL, which is etiologically associated with HTLV-I, most patients with CTCL are serologically negative for HTLV-I.11,12 Other investigators have found serologic evidence for Epstein-Barr virus and cytomegalovirus, but these findings have not been substantiated.15–15 Immunosuppression and/or immunosuppressive therapy might be a risk factor for the development of CTCL, as documented in patients after treatment with tumor necrosis factor-α (TNF-α) antagonists, treatment of organ transplant or of Hodgkin disease, and in human immunodeficiency virus (HIV)-positive patients.16–19 MF is the prototype of CTCL and the best-studied entity. MF classically presents as an indolent course, with slow progression over years or sometimes decades. The disease may evolve from erythematous patches to infiltrated plaques and eventually to tumors (Fig. 107-1). However, about 30% of patients have skin tumors (T3) or erythroderma (T4) only at initial presentation.20 Patients usually have a prolonged history of a rash in sun-protected areas, such as the lower abdomen, upper thighs, buttocks, inner upper arms, and breasts in women. Pruritus may or may not have be associated. The majority of patients remain in clinical stages limited to the skin; however, 20% of patients progress into more aggressive and advanced disease with either cutaneous or extracutaneous tumor manifestations, with an estimated 5-year survival rate of 25% to 40%. MF has numerous clinical and histologic variants. Besides the conventional type of MF, three distinct variants have been recognized in the 2005 WHO/EORTC classification, including folliculotropic MF, pagetoid reticulosis, formerly known as Worringer-Kolopp disease, and granulomatous slack skin, characterized by the development of lax skin folds (Fig. 107-2). Patients with folliculotropic MF have erythematous papules, plaques, and nodules with follicular involvement with or without alopecia; comedonal, acneiform, and cystic lesions; and associated intractable pruritus.21 Folliculotropic MF is frequently seen on the head and neck area in contrast to conventional MF. The clinical course of folliculotropic MF, particularly in early stages, is more aggressive; these patients respond poorly to topical therapy alone and require systemic therapy to achieve clinically meaningful responses. The 10-year progression-free survival is 45% compared with 91% in patients with conventional MF. Pagetoid reticulosis is a rare variant of MF with an indolent course and is predominantly seen in middle-aged men. Patients have a solitary psoriasiform plaque most often located on the lower extremity or foot. In contrast to classic MF, the epidermotropic atypical lymphocytes show a CD8+ phenotype by immunohistochemistry. Granulomatous slack skin is extremely rare and clinically characterized by indurated plaques resembling lax skin folds often in axillary and/or inguinal areas. On histology, there is a diffuse dermal infiltrate of giant cells and atypical lymphocytes with destruction of elastic fibers.22,23 Sézary syndrome (SS) is part of the spectrum of erythrodermic CTCLs as proposed by the International Society for Cutaneous Lymphomas (ISCL) (Table 107-2).24 Erythrodermic CTCLs can either develop de novo (as with SS) or as a progression of preexisting MF (as with erythrodermic MF). Patients who have erythrodermic MF may also have coexisting patches, plaques, or tumors. Three percent to 5 percent of all newly reported cases of CTCL are SS. SS is characterized by circulating, atypical, malignant T lymphocytes with cerebriform nuclei (Sézary cells), by the presence of erythroderma, and often by lymphadenopathy. Severe, disabling pruritus, ectropion, lymphoma-associated alopecia, palmoplantar keratoderma with fissures, and dystrophic nails are other common associated features (Fig. 107-3). Concomitant infections with dermatophytes or bacteria are common. Patients with SS are immunocompromised because of defective T-cell function and therefore predisposed for opportunistic skin infections such as colonization with Staphylococcus aureus.25 Table 107-2 Proposed Classification for Erythrodermic Cutaneous T-Cell Lymphoma (CTCL) and Relative Hematologic Criteria Devised by the International Society for Cutaneous Lymphoma (ISCL) MF, mycosis fungoides; NOS, not otherwise specified; TNMB, tumor, node, metastasis, blood. *B0, <5% circulating Sézary cells; B1, Sézary cell count of <1000 cells/m3 or <20% atypical T cells on peripheral smear; B2, Sézary cell count of >1000 cells/m3 or >20% atypical T cells on peripheral smear. From Vonderheid EC, Bernengo MG, Burg G, et al. Update on erythrodermic cutaneous T-cell lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 2002;46:95–106. A histopathological diagnosis of MF has proved to be difficult, because in initial and erythrodermic stages of disease inflammatory cells often predominate and may resemble benign dermatoses.26 It is not uncommon that patients may undergo multiple biopsies before a diagnosis can be rendered. A diagnostic algorithm for early MF was proposed by the ISCL and the Cutaneous Lymphoma Task Force of the EORTC that takes the clinical and histopathological findings into account (Table 107-3).27 Characteristic diagnostic features of classic MF/SS are a papillary dermal band–like infiltrate with atypical lymphocytes with hyperchromatic, hyperconvoluted nuclei, variable findings of inflammatory cells, and epidermotropism with infrequently seen Pautrier microabscesses (Fig. 107-4).28 Most MF/SS patients have the phenotype of the CD4+ T-helper (Th)/memory lymphocyte. Only a small number of cases, such as pagetoid reticulosis or hypopigmented variant of MF, are of the cytotoxic/suppressor T-cell subset expressing a CD8+ phenotype. SS patients also show commonly nonspecific histopathological findings, and a definitive diagnosis may be made by flow cytometry.29,30 It is important to differentiate tumor stage MF from non-MF subtypes of CTCL. In conventional MF, tumor lesions usually develop in the setting of patch/plaque disease and not de novo with the latter previously classified as tumor d’emblée type MF. It is now recognized that many of these cases represent non-MF cases and may fit best within the group of primary cutaneous PTCL, not otherwise specified. Table 107-3 Key Diagnostic Criteria for Mycosis Fungoides/Sézary Syndrome From Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007;110:1713–22. Immunophenotyping by immunohistochemistry or by flow cytometry and molecular studies have an important role in the evaluation of CTCL and aid into the diagnosis. There is no unique marker for the malignant lymphocyte, but an increased CD4 to CD8 ratio with an aberrant loss of T-cell lineage markers such as CD5, CD7, and/or CD26 is a common finding.29 A clonal gene rearrangement of the T-cell receptor can be detected in many cases and support the diagnosis using standard polymerase chain reaction (PCR) methods, but this has been complicated by the dilemma of clonality detected in benign skin disorders.* Furthermore, data on microsatellite DNA studies suggest that tumor cells may arise from multiple subclones and show a multiple-lineage progression.31 Accurate staging of patients with MF/SS is essential both for its prognostic value and for treatment decisions. The widely used and recommended staging system for CTCL relies on the TNMB (tumor, node, metastasis, blood) classification adopted by Bunn and Lamberg in 1979, which was revised by the ISCL and EORTC (Table 107-4).27,32 It considers the extent of skin involvement (T), presence of lymph node (N) and visceral disease (M), and detection of Sézary cells in the peripheral blood (B). Patients with patch/plaque disease are classified as either stage IA (T1, N0, M0, B0) with less than 10% of body surface area (BSA) involved or stage IB (T2, N0, M0, B0) with more than 10% BSA involved. By rule-of-thumb, the palm and digit of one hand represent 1% of BSA. Stage IIA disease (T1–2, N1, M0, B0) includes patients with skin findings of stage IA/IB disease with architectural preservation of any clinically abnormal lymph nodes. Currently, two histopathological grading systems, the Dutch and the National Cancer Institute/Veterans Administration (NCI-VA) classification system for lymph node evaluation, are used. Whereas the Dutch system is based on the presence of large cerebriform nuclei (>7.5 µm) and the degree of architectural effacement, the NCI-VA system uses the relative numbers of such cells in the paracortex of lymph nodes and nodal architecture to determine the extent of disease. Nodal involvement is characterized by partial (N2) or complete (N3) architectural effacement. Stage IIB (T3, N0–2, M0, B0) is associated with the development of skin tumors with or without associated lymph node involvement. The ISCL/EORTC has recommended definitions based on the degree of blood involvement for subsets of erythrodermic CTCL to address the differences among erythrodermic CTCL. A B1 rating is defined as a Sézary cell count of less than 1000 cells/m3 or less than 20% Sézary cells on peripheral smear. A B2 rating would be more than 1000 cells/m3 or greater than 20% Sézary cells on peripheral smear. Patients with erythroderma without significant lymph node involvement are stratified into stage IIIA (T4, N0–2, M0, B0) and stage IIIB (T4, N0–2, M0, B1) based on the presence of low blood tumor burden (B1) that allows for tracking of its prognostic significance in erythrodermic CTCL. High blood tumor burden (B2) is an independent prognostic variable as shown by others, and the B2 rating is now considered to be comparable to nodal involvement (N3).32a Stage IVA is therefore any skin stage with either blood (B2) or nodal disease (N3). Stage IVB is defined by any stage with visceral involvement (M1). The ISCL/EORTC considers splenomegaly even without biopsy confirmation as visceral disease. Suspected liver or bone marrow disease should be confirmed by biopsy. Table 107-4 TNMB Classification and Staging for Patients with Mycosis Fungoides/Sézary Syndrome BSA, body surface area; TNMB, tumor, node, metastasis, blood. Histologic classification of lymph node (LN) involvement is also used for prognostic value and for treatment decisions in patients diagnosed with MF/SS.33,34 It was found that the LN rating correlates with disease progression and survival.33,34 Moreover, detection of a monoclonal T-cell population within the lymph nodes is associated with an inferior survival and outcome regardless of the LN class.35 LN1 rating defines reactive changes, LN2 and LN3 nodes describe small or large clusters of atypical cells in paracortical T-cell regions, whereas LN4 nodes define frank effacement. The most important prognostic factors for survival remain the T stage (skin tumor burden), extracutaneous manifestation, and patient age.36 The Stanford experience with MF/SS patients demonstrates the impact of stage on overall survival (OS) rate (see Fig. 107-4).36 Although the OS rate of all patients with MF is 68% at 5 years and 17% at 30 years, the specific survival of patients ranges widely depending on T classification and stage at initial presentation. Patients with SS have an estimated 5-year survival rate of 24%. The largest study, consisting of 525 patients, showed an OS of 97% in patients with T1 (less the 10% body surface involvement) at 5 years, compared with 40% and 41% in T3 and T4 disease, respectively. In addition, several independent adverse prognostic factors have been identified, including large cell transformation, follicular mucinosis, thickness of tumor infiltrate, and increased LDH and β2-microglobulin levels.39–39 Patients with large circulating Sézary cells were also found to have a worse prognosis.40 A high Sézary cell count, loss of T-cell subset markers such as CD5 and CD7, and chromosomal abnormalities in T cells are also independently associated with a poor outcome.41 The existence of a blood clonal T-cell population, detected by PCR, was of poor predictive survival value, independent of the T stage and lymph node involvement.42 Recent multivariant analyses in survival outcomes and prognostic factors of MF and SS patients using the ISCL/EORTC revised staging proposal confirmed that the presence of a T-cell clone in blood (identical to the cutaneous T-cell clone) in the absence of morphologic evidence of blood involvement (B0b) was associated with a significantly worse OS rate and disease-specific survival (DSS) rate compared with those patients with no peripheral blood T-cell clone (B0a).43 In contrast, the presence of a high number of cytotoxic CD8+ T lymphocytes in the cutaneous infiltrate, as well as the density of epidermal Langerhans cells greater than 90 cells/mm2, is associated with a better prognosis.44,45 Large cell transformation is generally associated with a poor outcome. A recent study on prognostic factors in 100 patients with large cell transformed MF showed a median survival rate of 24 months (range, 1 to 235 months) with a 5-year DSS and OS rate of 38% and 33%, respectively.39 These results are similar to those from previous studies with median survival rates ranging from 12 to 36 months and 5-year OS rate from 11% to 32%.38,46–48 The most important prognostic factors included advanced stage at transformation, CD30 negativity, folliculotropic MF, and increased extent of skin lesions. There is no difference in outcome in patients with clinical stage IIB with or without large cell transformation. In contrast, another recent study of 1502 patients with MF/SS, including 70 patients with transformed MF at the time of first diagnosis, suggested a better prognosis of patients with LCT compared with all previous studies; the median survival was 8.3 years, and the 5-year OS and DSS rates were 63% and 65%, respectively.43 Current guidelines recommend a more aggressive approach to patients with large cell transformation49; however, the recent studies highlight that not all patients with large cell transformation may do worse. Most malignant T cells in CTCL are clonally derived from CD4+ CD45RO+ Th cells.50 In early stages of MF, the T-cell infiltrate consists of both malignant CD4+ and reactive CD8+ T cells with a dominance of Th1 cytokines such as interferon-γ (IFN-γ), interleukin (IL)-12, and IL-2.51 In later stages, there is a gradual increase of malignant CD4+ T cells, decrease in nonmalignant CD8+ T cells, and a shift to Th2 cytokine dominance (IL-4, IL-5, IL-10, and IL-13).52 These changes correlate with disease progression, host immunosuppression, and susceptibility to infection.53 Biological immune modifiers such as IFN-α, IFN-γ, and IL-12 are therapeutically effective in CTCL by stimulating Th1 cytokines and boosting host immune responses. An IL-17–producing T-cell (Th17) population in cutaneous lesions of MF/SS patients was also identified. Interestingly, the in vitro stimulation of MF/SS cells with IL-17 did not enhance their cell proliferation.54 In another study, IL-17 protein was found to be mediated by IL-2/IL-15 through the Janus-activated kinase (JAK3)/signal transducer and activator of transcription (STAT3) pathway.55 Whether distinct CTCL subtypes derive from Th1, Th2, or Th17 cells remains to be shown. Controversial results were found for programmed cell death (PD)-1 expression. Increased PD-1 expression has been shown on circulating CD4+ cells in SS patients when compared with MF patients, which could imply a role for an aberrant increase in PD-1 expression in the progression of tumors.56 Follicular Th cells display a distinct gene expression profile positive for PD-1, CXCL13, and BCL-6. Recent data showed increased PD-1 expression in pseudolymphoma and cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma, suggesting a follicular Th cell phenotype.57,58 Further research is needed in this area to determine whether the increase in PD-1 expression protects the tumor cells from elimination or if the increased PD-1 expression is a response of immunocompetent cells that are simply chronically stimulated by tumor antigens. The malignant T cells in MF express skin-homing markers of a skin-resident effector memory T cell, whereas circulating T cells in SS/erythrodermic CTCL express markers of a central memory T cell.59 These differences may explain their different clinical behavior and response to treatment. The malignant skin-resident T cells express the chemokine receptors (CCR)4 and CCR10 among others that are required for migration and homing into the skin. Malignant circulating T cells express CCR7 and L-selectin required for lymph node and peripheral blood. The chemokine ligand (CCL)17, a CCR4 ligand expressed on epidermal keratinocytes, endothelial cells, and dendritic cells, facilitates extravasation and migration of CTCL cells into the skin and epidermis. CCL27, a CCR10 ligand expressed on keratinocytes, has been implicated in both skin and nodal homing of CTCL cells.60 Anti-CCR4 is being evaluated in clinical trials including CTCL patients. Persistent activation of the neoplastic T cells is demonstrated by the constitutive phosphorylation of intracellular signaling protein STAT3.61 These cells may express the activation markers CD45RO and the IL-2α receptor (CD25) that has provided a target for biological therapy with denileukin diftitox.62 Naturally occurring regulatory T cells (Tregs; CD4+, CD25+, FOXP3+) also express the CD25 molecule. These cells suppress the activity of other immune cells, thus maintaining immunologic tolerance. Tregs appear to be dysregulated in CTCL.63 Early cutaneous MF lesions contain numerous FOXP3+ infiltrating Tregs that decrease in number in advanced lesions. The high frequency of FOXP3+ infiltrating Tregs may suppress tumor proliferation and has been correlated with improved survival.63 SS patients have very low levels of Tregs but high levels of malignant T cells expressing a Treg-phenotype (FOXP3+ CD25−). These malignant FOXP3+ Tregs express cytotoxic T-lymphocyte antigen (CTLA-4), IL-10, and transforming growth factor–β (TGF-β), which suppress immunity and diminish the antitumor response.53 CTLA-4 is a co-inhibitory molecule expressed on T cells that inhibits T-cell activation and proliferation and confers resistance against activation-induced cell death.64,65 Tregs also constitutively express CTLA-4, which is necessary for their functioning to maintain peripheral tolerance and to prevent autoimmunity.66,67 High CTLA-4 expression was found in peripheral blood mononuclear cells from patients with MF, and higher expression levels correlate with increased tumor burden. Th1-derived cytokines such as IL-2 and IFN-γ upregulate expression of CTLA-4.68 Whether increased CTLA-4 expression relates to Treg-like properties that CTCL cells acquire during disease progression is not clear. Malignant CD4+ T cells from cutaneous lesions and peripheral blood samples in MF and SS have decreased and/or defective Fas expression and decreased Fas expression has been correlated with more aggressive disease as well as resistance to Fas-mediated apoptosis.69–72 Downregulation of Fas in CTCL occurs through multiple mechanisms: mutations in the Fas gene, production of nonfunctioning splice variants, and promoter hypermethylation.70,73 In this context, malignant T cells in CTCL may acquire resistance to FasL signaling through the increased expression of cFLIP, an intracellular apoptosis inhibitor.69 T cells generally express BCL-2, which inhibits apoptosis and is widely and stably expressed in all stages of MF.74 STAT3 directly regulates BCL-2/BAX genes involved in apoptosis.75 Data suggested that inhibition of STAT3 signaling in CTCL cells through the JAK inhibitor Ag490 induced apoptosis through decreased expression of antiapoptotic BCL-2 and increased expression of pro-apoptotic BAX protein.76 Improvements in microarray technology and computational analysis of genomic data have led to discoveries of underlying chromosomal mutations in tumor suppressor and oncogenes involved in CTCL.79–79 Chromosomal regions with significant gains include 8q (including the MYC oncogene), 17q, and 10p13 (including GATA3, a transcription factor that promotes Th2 cytokine production).79 Additionally, a recent study suggested that amplifications on 4q12 (including KIT), 7p11.2 (including EGFR), and 17q25.1 may be highly associated with patients refractory to treatment.79 Patients with large cell transformation show chromosomal clonal evolution with changes in ploidy levels and structural aberrations.80 Specific oncogenes have been examined for defining new prognostic factors in CTCL. Deletions have been found on chromosomes 17p (including TP53), 10p and 10q (including PTEN and FAS), 13q including RB1, and 9p21.3 (including CDKN2A).79 Expression profiles showed upregulation of genes involved in the TNF signaling pathway and decreased expression of tumor suppressor genes such as TGF-β receptor II whereas EPHA4 and TWIST were overexpressed.81,82 Aberrant expression of STAT4, GATA3, T-plastin, CD1D, TRAIL, CDO1, and DNM3 was also found and may serve as potential targets for novel treatment strategies.83,84 Evidence of epigenetic silencing of hypermethylation of individual genes or gene-specific promoters associated with tumor progression have been observed in more advanced stages of CTCL.31,85 In CTCL, promoter hypermethylation leads to dysregulation of cell cycle (CDKN2B, CDKN2A, TP73), apoptosis (TMS1, TP73), DNA repair (MGMT), chromosomal instability (CHFR), and microsatellite instability (MLH1) genes and proteins.85–89 Promoter hypermethylation of CDKN2B, CDKN2A, and MLH1 were found in both early and advanced stages of MF and SS, suggesting that early epigenetic alterations were responsible for the inactivation of these genes.89–89 The multilineage progression of CTCL is also supported by microsatellite instability, as determined by microdissected analyses of tumor infiltrates.31 Several studies have found microRNA (miRNA) signatures in CTCL, but data were inconsistent and distinct genetic markers for diagnosis and prognostication were not identified.90,91 More recently, a study has shown that miRNA classifier can distinguish CTCL from benign skin disorders.92 A microarray screen found that five miRNAs (miR-203, miR-205, miR-326, miR-663b, and miR-711) distinguish CTCL from benign skin diseases with accuracy greater than 90%.93 Interestingly, these five miRNA classifiers were not affected by drug treatment. In tumor stage MF, miR-93, miR-92A, and miR-155 were upregulated in comparison with benign inflammatory skin diseases.94 In SS, most miRNAs were downregulated but miR-21, miR-486, and miR-214 are upregulated and involved in apoptotic resistance.95
Cutaneous T-Cell Lymphoma and Cutaneous B-Cell Lymphoma
Introduction
WHO-EORTC
Frequency (%)
5-Year Survival (%)
CUTANEOUS T-CELL AND NK-CELL LYMPHOMA
INDOLENT
Mycosis fungoides
44
88
Follicular mycosis fungoides
4
80
Pagetoid reticulosis
<1
100
Granulomatous slack skin
<1
100
CD30+ LYMPHOPROLIFERATIVE DISORDERS
Anaplastic large cell lymphoma
8
95
Lymphomatoid papulosis
12
100
Subcutaneous panniculitis–like T-cell lymphoma
1
82
CD4+ small/medium pleomorphic T-cell lymphoma
2
72
AGGRESSIVE
Sézary syndrome
3
24
Cutaneous peripheral T-cell lymphoma, unspecified
2
16
Cutaneous aggressive CD8+ T-cell lymphoma
<1
18
Cutaneous γ/δ T-cell lymphoma
<1
Cutaneous NK/T-cell lymphoma, nasal-type
<1
CUTANEOUS B-CELL LYMPHOMA
Indolent
Follicle center cell lymphoma
11
95
Marginal zone lymphoma
7
99
Intermediate Clinical Behavior
Large B-cell lymphoma of the leg
4
55
Cutaneous diffuse large B-cell lymphoma, other
<1
50
Intravascular large B-cell lymphoma
<1
65
Epidemiology
Etiology
Cutaneous T-Cell Lymphoma
Mycosis Fungoides and Sézary Syndrome
Erythrodermic CTCL
Preexisting MF
Blood Findings
TNMB Classification
Sézary syndrome
Rarely
Leukemic
T4, N0–3, M0–1, B2*
Erythrodermic MF
Always
Absent or minimal
T4, N0–3, M0–1, B0–1*
Erythrodermic CTCL, NOS
Absent
Absent or minimal
T4, N0–3, M0–1, B0–1*

Criteria
Major (2 points)
Minor (1 point)
CLINICAL
Persistent and/or progressive patches/plaques plus:
Any 2
Any 1
Sun-protected areas
Size/shape variation
Poikiloderma
HISTOPATHOLOGICAL
Superficial lymphoid infiltrate plus:
Epidermotropism without spongiosis
Both
Either
Lymphoid atypia
Molecular
Clonal TCR rearrangement
Present
Immunophenotypic
CD2, CD3, CD5 < 50% of T cells
Any 1
CD7 < 10% T cells
Epidermal discordance from CD2, CD3, CD5, or CD7 phenotype of dermal T cells
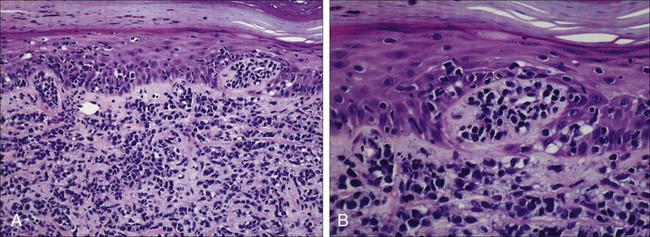
Staging and Prognosis
T (SKIN)
T1
Limited patch/plaque (<10% of BSA)
T2
Generalized patch/plaque (>10% of BSA)
T2a = patch only
T3
One or more tumors (>1 cm)
T4
Generalized erythroderma (>80% of BSA)
N (NODES)
N0
No clinically abnormal peripheral lymph nodes
N1
Clinically abnormal peripheral lymph nodes
Dutch grade 1, NCI grade LN0–2
N1a
Clone negative
N1b
Clone positive
N2
Clinically abnormal peripheral lymph nodes
Dutch grade 2, NCI grade LN3
N2a
Clone negative
N2b
Clone positive
N3
Clinically abnormal peripheral lymph nodes
Dutch grade 3, NCI grade LN4, clone positive or negative
Nx
Clinically abnormal peripheral lymph nodes
No histologic confirmation
M (VISCERA)
M0
No visceral organ involvement
M1
Visceral organ involvement (pathology confirmation required)
B (BLOOD)
B0
Atypical circulating cells not present (<5%)
B0a
Clone negative
B0b
Clone positive
B1
Atypical circulating cells present (>5%)
B1a
Clone negative
B1b
Clone positive
B2
≥1000/µL Sézary cells, clone positive
Stage
T
N
M
B
IA
1
0
0
0,1
IB
2
0
0
0,1
IIA
1,2
1,2
0
0,1
IIB
3
0-2
0
0,1
IIIA
4
0-2
0
0
IIIB
4
0-2
0
1
IVA1
1-4
0-2
0
2
IVA2
1-4
3
0
0-2
IVB
1-4
0-3
1
0-2

Transformed Mycosis Fungoides/Sézary Syndrome
Biological Properties
Immunopathogenesis
Molecular Pathogenesis
Cutaneous T-Cell Lymphoma and Cutaneous B-Cell Lymphoma