Cushing’s syndrome describes the signs and symptoms associated with prolonged exposure to elevated levels of endogenous or exogenous glucocorticoids. This can be caused by taking glucocorticoid drugs, or by diseases that result in excess cortisol, adrenocorticotropic hormone (ACTH), or corticotropin-releasing hormone (CRH) levels.1,2
Cushing’s syndrome most commonly arises from iatrogenic causes when glucocorticoids have been used as anti-inflammatory and/or immunosuppressive treatment. The pathogenetic mechanisms of endogenous Cushing’s syndrome can be divided into ACTH dependent (80%) and ACTH independent (20%).
The most common (80%) ACTH-dependent type, known as Cushing’s disease, is caused by ACTH-secreting pituitary adenomas.1–3 In 1932, Harvey Cushing first described the presence of basophilic pituitary adenomas as a cause of Cushing’s disease.4 The remaining 20% of ACTH-dependent forms are due to nonpituitary ectopic sources of ACTH, such as the paraneoplastic manifestations of oat cell carcinoma, small-cell lung carcinoma, or a carcinoid tumor. When ACTH is ectopically secreted by small cell carcinoma of the lung or widely metastatic cancer, patients can have a rapid and sever onset of Cushing’s syndrome.1,2,5
Approximately 20% of Cushing’s syndrome is ACTH independent, resulting in excess of cortisol secretion by adrenocortical tumors. In the majority of cases, these tumors are unilateral and 60% are benign adenomas and 40% are adrenal carcinomas. Rarely, ACTH-independent Cushing’s syndrome is caused by bilateral macronodular hyperplasia, primary pigmented nodular adrenocortical disease (which may be a part of Carney complex), or McCune–Albright syndrome.1–3,5
Exogenous corticosteroid exposure is the most common cause of Cushing’s syndrome. More than 10 million Americans receive pharmacologic doses of glucocorticoids each year; however, there is no data as to the exact number of patients with exogenous Cushing’s syndrome.6
Previously, endogenous Cushing’s syndrome was thought to be a rare disorder with an incidence of approximately two to three cases per million per year. However, recent data have demonstrated an incidence of Cushing’s syndrome between 2% and 5% in obese patients with type 2 diabetes with poor blood glucose control and hypertension.7–9 Cushing’s syndrome caused by disease of the adrenal glands occurs four times more commonly in women than in men. Cushing’s disease (pituitary tumor secreting ACTH) has a 3.5 to 1 female-to-male predominance. No ethnic disparities are identified. Most cases are diagnosed in adults between the ages of 20 and 50, although it can occur at any age. Pediatric cases are unusual but well documented.1
Patients with uncontrolled Cushing’s syndrome have a fivefold increased risk in mortality.10,11 Risk factors for developing endogenous Cushing’s syndrome are being delineated as more is learned about the disease. There is a genetic predisposition in patients with Carney complex, McCune–Albright syndrome and multiple endocrine neoplasia type 1 (MEN1) syndrome.1 Recent work has also associated obese patients with poorly controlled diabetes at having an increased risk of a hypercortisol states.
Recent work in the Cushing’s syndrome pediatric population studied bone age development and found that contrary to previous beliefs, bone age maturation is advanced, not delayed in the majority of pediatric patients. Prolonged hypercortisol, insulin resistance, obesity, and elevated androgens contributed to skeletal maturity and could lead to compromised growth potential. Surgical correction of either Cushing’s disease or the adrenal source showed a normalcy of bone age and allowed for children to potentially reach normal height. Early recognition and proper management of Cushing’s syndrome in children is crucial to correct not only bone age abnormalities but also their metabolic profile.12
There has been work into the molecular pathways causing Cushing’s syndrome. Several studies have looked at specific genes associated with hypercortisolism. Recently, a mutation in ARMC5 was discovered in a subset of patients with macronodular hyperplasia.13 Mutations in the PRKAR1A gene have been associated with primary pigmented nodular adrenocortical disease (PPNAD) and work recently published demonstrates phenotypic differences in fat distribution among Cushing’s syndrome patients with and without PRKAR1A mutations.14–16 Thymic carcinoids secreting ACTH in patients with MEN1 have been reported to cause Cushing’s syndrome and patients with MEN1 and thymic carcinoids are encouraged to be screened for Cushing’s syndrome.17,18 Most recently, a point mutation has been identified in the human glucocorticoid receptor gene causing glucocorticoid resistance.19
In Cushing’s syndrome, the hypothalamic–pituitary–adrenal axis pathway becomes dysregulated. The hypothalamic–pituitary–adrenal axis (HPA or HTPA axis), also known as the limbic–hypothalamic–pituitary–adrenal axis (LHPA axis), is a complex set of direct signals and feedback interactions among the hypothalamus, the pituitary gland, and the adrenal glands.20
The paraventricular nucleus of the hypothalamus contains neuroendocrine neurons that synthesize and secrete vasopressin and CRH. CRH and vasopressin are released from neurosecretory nerve terminals at the median eminence of the hypothalamus. CRH is transported to the anterior pituitary through the portal blood vessel system of the hypophyseal stalk and vasopressin is transported by axonal transport to the posterior pituitary. Once there, CRH and vasopressin act synergistically to stimulate the secretion of stored ACTH from corticotrope cells. ACTH is transported by the blood to the adrenal cortex of the adrenal gland, where it rapidly stimulates biosynthesis of corticosteroids including cortisol. Glucocorticoids in turn act back on the hypothalamus and pituitary to suppress CRH and ACTH production in a negative feedback cycle.5,21 Cortisol acts on two types of receptor in the brain, mineralocorticoid and glucocorticoid receptors, and these are expressed by many different types of neurons. An important target of glucocorticoids is the hypothalamus, which is a major controlling center of the HPA axis.22
Although Cushing’s disease is the most common form of endogenous Cushing’s syndrome, very little is known about the pathogenesis of pituitary tumors.23 In general, pituitary tumors have been characterized as expressing low levels of cyclin-dependent inhibitor p27, overexpression of cyclin E, and a high ki67 proliferation index.24,25 About 90% of pituitary tumors express CRH-1 receptor.26 Pituitary or corticotrope tumors also overexpress the proopiomelanocortin (POMC) gene which codes for the peptide product that is cleaved to become corticotropin.27 Recent work has demonstrated a loss of corticotropin receptor expression, increased cortisol inactivation by 11-beta hydroxysteroid dehydrogenase, and reduced expression of bridging protein which is a protein associated with glucocorticoid feedback.1,28 A study of a small cell lung carcinoma cell line, DSM-79, has shown that POMC is activated by transcription factors that are distinct from transcription factors found in the pituitary tumors and bind the promoter in an unmethylated state.29 Carcinoid tumors are thought to have a molecular profile similar to pituitary adenomas.30
More is known about ACTH-independent adrenal Cushing’s syndrome. Macronodular adrenal hyperplasia has been characterized as having both aberrant receptor expression as well as overexpression of receptors normally found in adrenal tissue.31 These receptors include gastric inhibitory peptide, vasopression, interleukin 1, luteinizing hormone, leptin, and serotonin. Activation of receptors coupled to cyclic AMP is thought to cause hyperplasia. Aberrant receptors also occur in unilateral adenomas but to a much lesser degree.31
Cortisol is a major stress hormone that has effects on many tissues in the body, including the brain. The clinical manifestations of Cushing’s syndrome are therefore variable and can differ in severity depending on the degree and duration of hypercortisolism. The classical phenotype of Cushing’s syndrome includes cardiovascular, metabolic, dermatological, musculoskeletal, and psychiatric manifestations. However, in many cases, patients may have only isolated symptoms.32
Symptoms include rapid weight gain, truncal obesity, cervical fat pad growth, and moon facies. The limbs are usually spared from weight gain. Other symptoms include hyperhidrosis, telangiectasias, thinning of the skin which leads to bruising and dryness, and purple or red striae. The striae are a result of rapid weight gain in the setting of stretched and thinned skin which is susceptible to hemorrhage. Patients may also complain of proximal muscle, hirsutism or baldness, and/or extremely dry and brittle hair. In rare cases, Cushing’s syndrome can cause hypocalcemia. High cortisol levels may also lead to insomnia, reduced libido, impotence in men, amenorrhea/oligomenorrhea, and infertility in women.33–36
Cognitive conditions such as memory loss and attention dysfunctions are associated with elevated cortisol levels. Patients may suffer various psychological disturbances, ranging from depression to euphoria to psychosis.1,33
Other signs include polyuria, insulin resistance, and persistent hypertension. Insulin resistance may cause diabetes mellitus. Left untreated, Cushing’s syndrome can lead to heart disease and increased mortality. Excess cortisol can also cause increased secretion of mineralocorticoids. High concentrations of mineralocorticoids can worsen hypertension and cause electrolyte imbalances. Additionally, prolonged exposure to high levels of cortisol may lead to impaired wound healing related to cortisol’s suppression of the immune and inflammatory responses. Osteoporosis is also an issue in Cushing’s syndrome due to the fact that osteoblast activity is inhibited.33–37
The diagnosis of Cushing’s syndrome due to endogenous overproduction of cortisol requires the demonstration of inappropriately high serum or urine cortisol levels. This can be done utilizing one of the following tests. Two abnormal results establish the diagnosis of Cushing’s syndrome.38
The 24-hour urinary free cortisol (UFC) determination can be used as initial screening tool for Cushing’s syndrome. Valid results depend on adequate urine collection. UFC values higher than three to four times the upper limit of normal are highly suggestive of Cushing’s syndrome. Values that are higher than the normal reference range but less than three to four times the upper limit of normal are considered inconclusive and require additional testing. Patients that have Cushing’s syndrome may have UFC values that fall into the normal range. At least two measurements are required to make a diagnosis. Three UFC levels in the normal range exclude the diagnosis of endogenous Cushing’s syndrome.38–40
The salivary cortisol test is performed by collecting saliva between 11 p.m. and midnight. Saliva is easily collected and cortisol is stable in saliva at room temperature for several days. This method of collection can be performed by the patient at home. It is a good test for patients suspected of having cyclical or intermittent Cushing’s syndrome, as these patients can collect multiple samples over several days at home by the patients. Cotton pledgets collect saliva and then are stored for analysis. All samples may be returned at once. At least three samples from three different days with elevated salivary cortisol levels are required for a diagnosis of Cushing’s syndrome.41,42
The dexamethasone suppression test is dependent on the normal physiology of the hypothalamic–pituitary–adrenal axis. Glucocorticoids inhibit secretion of hypothalamic CRH and pituitary ACTH but do not directly affect adrenal cortisol production. The production of cortisol is controlled by ACTH and therefore decreases in ACTH lead to decreases in plasma and urine cortisol. The overnight 1-mg dexamethasone suppression test requires administration of 1 mg of dexamethasone at 11 p.m. with the subsequent measurement of cortisol at 8 a.m. In unaffected individuals, the plasma cortisol level will be less than 1.8 μg/dL.43–46
For patients with inconclusive results or those that have high clinical suspicion but equivocal results in other screening methods, the dexamethasone-CRH test is used. It is intended to distinguish patients with Cushing’s syndrome from those with pseudo-Cushing states. The dexamethasone-CRH test combines a 48-hour low-dose dexamethasone suppression test with CRH stimulation. Low-dose dexamethasone is given every 6 hours starting at around 8 a.m. CRH is given 6 hours after the last dose of dexamethasone. Serum cortisol and ACTH levels are drawn at 15-minute intervals for 1 hour after CRH administration. A cortisol level greater than 50 nmol/L (1.4 μg/dL) indicates Cushing’s syndrome (see Fig. 42-1).47,48
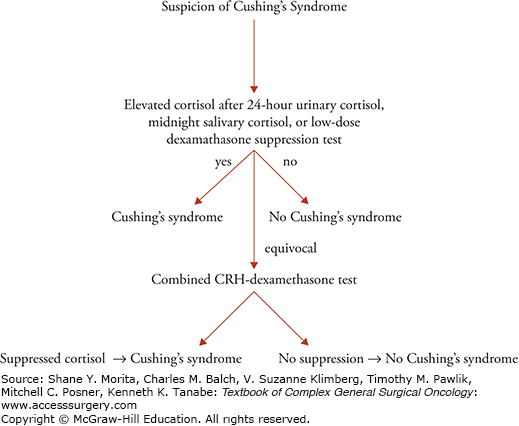
False-positive results can occur when testing for Cushing’s syndrome. Obesity, alcoholism, eating disorders, strenuous exercise, and chronic renal failure may all give false-positive results.48 Medications may interfere with both cortisol binding and metabolism of dexamethasone and thus result in falsely elevated cortisol levels. Estrogen and tamoxifen increase corticosteroid-binding globulin, which may cause increases in serum cortisol levels. Medications such phenytoin, phenobarbital, and rifampin accelerate the metabolism of dexamethasone by the induction of CYP3A4.46 Additionally, acute illness activates the HPA axis, resulting in increases in ACTH and cortisol. The laboratory workup for Cushing’s syndrome should not be performed when patients are acutely ill.
Once the diagnosis is established, the next step requires determining the etiology of Cushing’s syndrome. The first step involves identifying if the hypercortisolism is an ACTH-dependent or ACTH-independent disorder. This is done by measuring serum ACTH.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


