Fig. 38.1
Key time points in the discovery and development of immuno-oncology drugs for the treatment of hematologic cancers
38.2.1 The Famous CD20 Story
Overcoming the barriers of xenogeneic immunogenicity through humanization of murine antibodies led to the development of rituximab, the first humanized mAb directed against CD20, which showed unprecedented clinical activity in relapsed CD20 diffuse large B-cell lymphoma [3–5]. In the landmark randomized clinical trial published in 2002, the French GELA group demonstrated that the addition of rituximab to the standard CHOP regimen was able to increase the response rate and prolong event-free and overall survival (OS) up to 10 years (44% vs 28%, respectively) in elderly patients with diffuse large B-cell lymphoma [6]. Since that pivotal trial, rituximab has proven its efficacy in nearly all types of CD20-positive B-cell malignancies and now represents the standard of care in indolent and aggressive non-Hodgkin lymphomas (NHL) as well as chronic lymphocytic leukemia (CLL). Recently, the results of two large randomized clinical trials demonstrated the superiority of rituximab-based strategies in Burkitt’s lymphoma and B-lineage adult acute lymphoblastic leukemia (ALL) [7, 8].
Second-generation mAb against CD20, obinutuzumab (GA101), is a glycoengineered type II humanized mAb that was designed to have a stronger affinity to the FcγRIII on immune cells and potentially being superior to rituximab in vitro. As compared to rituximab, obinutuzumab demonstrated more direct cell death and enhanced ADCC/ADCP but lower CDC. In addition, obinutuzumab, which binds to a single CD20 tetramer, is believed to get less internalized than rituximab, which might increase ADCC/ADCP. Obinutuzumab was approved for the treatment of CLL in elderly or unfit patients following the results of a large randomized trial that demonstrated the superiority of chlorambucil and obinutuzumab over chlorambucil and rituximab in terms of response rate and complete response [9]. It should be noted that obinutuzumab plus chlorambucil was associated with more toxicities. In CLL, a combination of obinutuzumab with novel agents such as the bcl-2 antagonist venetoclax and the btk inhibitor ibrutinib is being evaluated. Promising preliminary results presented in conferences make it likely that obinutuzumab will be widely used in CLL in the near future.
Furthermore, obinutuzumab has been recently approved in relapsed follicular lymphoma in combination with bendamustine based on longer progression-free survival (PFS) when compared to bendamustine monotherapy in the GADOLIN phase 3 clinical trial [10]. Similarly, the primary results of the GALLIUM study showed that obinutuzumab-based induction and maintenance increased PFS in patients with previously untreated follicular lymphoma [11]. However, the success observed in follicular lymphoma and CLL might not be valid in more aggressive lymphomas. Indeed, the GOYA study, a large randomized phase III trial that compared head-to-head rituximab or obinutuzumab plus CHOP in 1418 patients with untreated diffuse large B-cell lymphoma, failed to meet its primary endpoint (investigator-assessed PFS) [12].
38.2.2 Targeting Molecules Other than CD20
38.2.2.1 Alemtuzumab
Alemtuzumab is a mAb directed against CD52, a cell surface antigen expressed by B cells, T cells, and monocytes. It was originally approved as a single agent for the treatment of fludarabine-refractory and del17p CLL [13]. Although alemtuzumab appeared effective in one-third of patients with fludarabine-refractory disease, its broad immunosuppressive activity was associated with severe and lethal opportunistic infections [14]. In CLL and other subtypes of lymphomas, the results of most clinical trials evaluating the association of alemtuzumab with chemotherapy failed to demonstrate a clear advantage over standard therapy, with some studies being prematurely interrupted because of an alarmingly higher death rate related to infections. For the same reason, its use in conditioning regimen in non-myeloablative allogeneic HSCT has been limited by an increased risk of life-threatening infectious complications and a higher rate of disease relapse [15]. Commercialization of alemtuzumab for hematological malignancies was interrupted in 2012, because the manufacturer wished to rebrand the product under a different name for multiple sclerosis. It remains accessible through specific distribution program for CLL in different countries.
38.2.2.2 Gemtuzumab Ozogamicin
Gemtuzumab ozogamicin (GO) is a humanized mAb directed against the CD33 surface antigen coupled to calicheamicin, a potent antitumor anthracycline antibiotic. In AML, CD33 represents an interesting target because it is expressed by the majority of AML cells. Following its approval by the FDA after the promising phase 2 data in relapsed older adults with AML, GO has been the subject of controversies and was even withdrawn following the results of a large randomized trial (recently reviewed by Rowe and Lowenberg) [16, 17]. However, aggregated data from four randomized trials have renewed the interest of using GO in newly diagnosed AML, particularly in patients with more favorable cytogenetic. In addition, GO seems particularly active in newly diagnosed and relapsed acute promyelocytic leukemia, which expresses high level of CD33 [18]. Given the absence of novel effective agents in AML over the last three decades, the use of GO should probably be reappraised in the light of these results.
38.2.3 Newer Target of Interest
38.2.3.1 Brentuximab Vedotin
Brentuximab vedotin (BV) is an antibody drug conjugate consisting in a mAb directed against CD30 conjugated to the microtubule-disrupting agent MMAE via a protease-cleavable linker. CD30 is a cell surface molecule belonging to the tumor necrosis factor (TNF) receptor superfamily that is mainly expressed by subpopulations of B and T cells upon activation [19]. The expression of CD30 has been reported in various hematologic and solid cancers. In hematology, the principal malignancies expressing CD30 are classical Hodgkin lymphoma (cHL), anaplastic large cell lymphoma (ALCL) anaplastic large cell lymphoma, primary mediastinal B-cell lymphoma, T-cell lymphomas, and EBV-induced posttransplant lymphoproliferative disorders.
BV demonstrated clinical activity in relapsed/refractory CD30-positive lymphomas [20] and is approved as a single agent for this condition and in ALCL. In HL, administered in monotherapy, BV induces an overall response rate (ORR) of 75% and is associated with durable responses in nearly half the patients relapsing after autologous SCT [21–23]. A phase 2 trial evaluating frontline BV as a monotherapy in older patients unfit for conventional chemotherapy showed a 92% overall response rate including 73% of complete remission [24].
Combination of BV with chemotherapy and radiotherapy was feasible and well tolerated in numerous phase I and phase II trials. A word of caution: BV should not be used in combination with bleomycin-containing regimen because of a high rate of pulmonary complications [25]. In a phase II pilot study evaluating BV and AVD followed by involved-site radiotherapy in early-stage unfavorable risk HL, 90% (26/29) and 93% (27/29) of patients achieved a negative PET scan after two and four cycles, respectively. A large phase III trial is currently evaluating BV plus AVD versus ABVD in advanced classical HL (NCT01712490). The AETHERA study examined the potential of BV administered as consolidation therapy post-autologous SCT in relapsed HL [26]. Median PFS was 42.9 months for patients in the brentuximab vedotin group compared with 24.1 months for those in the placebo group, supporting its efficacy in the posttransplant setting. Updated data confirm the superiority of the BV arm in the AETHERA study [27]. A limited number of studies have shown BV to control disease before [28] or in patients relapsing after allogeneic HSCT [29–31]. Altogether, BV has demonstrated a very promising activity in HL. The results of several ongoing phase III randomized trials, which are evaluating BV in addition with chemotherapy in either untreated or relapsed HL, might lead to the incorporation of BV in standard-of-care regimen for cHL.
BV has been prescribed for other CD30-positive lymphoid malignancies in small trials or case reports; therefore, data are less robust than with cHL. BV induced an 85% ORR with a median duration of 12.6 months in 58 patients with relapsed or refractory systemic anaplastic large cell lymphoma [32]. A phase I/II study of frontline BV in combination with chemotherapy in CD30-positive primary mediastinal B-cell lymphomas (PMBCL), diffuse large B-cell lymphomas (DLBCL), and gray-zone lymphomas is recruiting patients (NCT01994850). Further studies are needed to define its efficacy, especially in T-cell lymphomas where improvement is eagerly awaited given their usual resistance to standard chemotherapy.
38.2.3.2 Daratumumab
Daratumumab is a human IgG1 monoclonal antibody that targets CD38, a transmembrane glycoprotein that is expressed by a large array of cell types including T cells, B cells, monocytes, and NK and NK/T cells. CD38 is ubiquitously expressed on multiple myeloma (MM) cells, which makes it an interesting therapeutic target. Recent experimental evidence suggests that the biological effect of daratumumab expands beyond the classical properties of therapeutic mAbs and may involve immunomodulation [33]. Daratumumab can deplete regulatory immune cells like myeloid-derived suppressor cells (MDSC), regulatory B cells, and a newly identified subset of CD38hi regulatory T cells, which allows expansion of CD4+ Th cells and CD8+ cytotoxic T cells and increased IFN-γ. Daratumumab as a single agent demonstrated very promising activity in two clinical studies in heavily pretreated patients with relapsed/refractory MM [34, 35]. Daratumumab induced remarkable response rates, including stringent complete responses, and prolonged clinical responses. These results led to its approval in MM with ≥3 lines of treatment including a proteasome inhibitor and an IMID or refractory to both.
Early data from the CASTOR and POLLUX trials, which evaluated the association of daratumumab with bortezomib and dexamethasone or lenalidomide and dexamethasone, respectively, in previously treated MM patients, have been published [36, 37]. Although the results of these studies are somehow limited by their short follow-up (7.5 and 13.5 months), both already showed a significant improvement in terms of response and PFS. The 12-month rates of PFS were 60.7% in the daratumumab group versus 26.9% in the control group (CASTOR) and 83.2% in the daratumumab group, as compared with 60.1% in the control group (POLLUX). Daratumumab has been rapidly moved to the frontline and is being evaluated in combination with other anti-myeloma agents. In HSCT-eligible MM patients, the large randomized phase III CASSIOPEA trial from the IFM is comparing the standard-of-care regimen VTD (bortezomib, thalidomide, and dexamethasone) to daratumumab plus VTD (NCT02252172). In older patients, VMP (bortezomib, melphalan, and prednisone) is challenged against daratumumab plus VMP (NCT02195479). If the results of these studies confirm its efficacy in MM, daratumumab might well become “the rituximab of myeloma.”
Besides MM, there is a rationale to try daratumumab in other hematological malignancies that express CD38. Preclinical data support evaluating daratumumab in high-risk CLL, in which the malignant B-cell population frequently expresses CD38 [38]. Recently, daratumumab was used with success in a case of refractory CD38+ NK/T-cell lymphoma [39].
38.2.3.3 Elotuzumab
Elotuzumab is another mAb directed against CS1/SLAMF7 receptor. SLAMF7 is uniformly expressed at a high level by both normal and malignant plasma cells, whereas other cell types to the exception of NK cells do not express it. Elotuzumab’s mechanism of action is different than other mAbs like daratumumab. Elotuzumab binds to SLAMF7 expressed by NK cells leading to their activation. Activated NK cells then kill elotuzumab-tagged plasma cells through engagement of CD16 with its Fc region.
Elotuzumab has little or no effect as a single agent in patients with relapsed/refractory MM [40]. However, it demonstrated activity in combination with other anti-myeloma drugs in phase I trials [35, 41]. In a randomized open-label phase 2 study, the addition of elotuzumab to bortezomib slightly increased the PFS in 150 previously treated MM patients [42]. The 1-year PFS rate was 39% vs 33% in the control arm, and in the updated analysis, the 2-year PFS rate was 18% vs 11%. The large randomized multicenter phase III ELOQUENT-2 trial compared the efficacy and safety of lenalidomide and dexamethasone with or without elotuzumab in 646 relapsed/refractory MM patients after ≤3 lines of treatment [43]. The ORR was 79% vs 66% in the control group, and median PFS was 19.4 months vs 14.9 months in the elotuzumab group and the control group, respectively. These results led to its FDA approval in 2015. In conclusion, elotuzumab in combination with lenalidomide has demonstrated clinical activity in MM, although its efficacy appears less impressive than daratumumab. Further studies are needed to clearly define the place of elotuzumab in the armamentarium against MM, in particular what should be the optimal combination.
38.2.3.4 Bispecific T-Cell Engagers
The previous sections revealed that compared to standard chemotherapy, mAbs offer a more selective and less toxic therapeutic approach. However, the major drawback of mAbs is the inability to recruit cytotoxic T lymphocytes to the tumor bed and transform non-inflamed tumor to inflamed one. Several lines of evidence have demonstrated that the number of tumor-infiltrating T cells directly correlates with the clinical outcome in different cancers [44]. It took more than one decade from the development of rituximab to envision methods capable of directly increasing and priming specific CTL within the tumor microenvironment. Two distinct strategies that illustrate this paradigm shift and successfully translate into the clinic are:
- 1.
Unleashing the priming and effector phase of T lymphocyte-mediated immune responses by suppressing the interaction of inhibition receptors and ligands with ICBs such as PD-1 and CTLA-4 blockades.
- 2.
Bypassing the exhausted T cells with chimeric antigen receptor T cell (CAR T cells): adoptive injection of transduced autologous T cells with a specific antigen-binding domain coupled to intracellular receptor on T cell thereby redirects cytotoxic T lymphocytes to specifically recognize cancer cells.
To overcome the cost, complexity, and delay of manufacturing personalized CAR T cells for each patient in a good manufacturing practice (GMP) facility, a different approach to redirect T cells was needed. From this perspective, the development of BsAbs represents a strategy of paramount interest. BsAbs also called dual-targeting antibodies have the capability to bind two different targets bringing two cells in contact. This technology allows engaging a patient’s own cytotoxic T cells to cancer cells and promotes a sustained tumor lysis. Following the clinical benefit in ALL, BsAbs represent one of the fastest-growing class of anticancer therapeutics, and more than 50 BsAbs are currently in clinical development [45, 46].
Bispecific T-cell engagers (BiTe) like blinatumomab consists in two scFv separated by a flexible and short linker (Fig. 38.2). The N-terminal scFv recognizes the tumor-associated antigens (TAA), and the C-terminal scFv binds to invariant CD3ε monoclonal antibody. The two arms are connected by a non-immunological linker and provide a remarkable flexibility. This therapeutic architecture is independent from TCR specificity and from peptide antigen presentation. Therefore, by bypassing the MHC I TCR axis on CTL, polyclonal T cells can be recruited to the tumor microenvironment and overcome the downregulation of MHC molecules that is often found on cancer cells. In vitro experiments revealed that BsAb like blinatumomab (CD3/CD19) coculture triggers immunological synapse between effector T and leukemic cells. These synapses allow the release of perforin and granzyme (pore-forming protein) by T cells further contributing to the cell nuclear condensation and membrane blebbing. The immunological synapses also lead to the upregulation of surrogate immune-activating receptors like CD25 and CD69 and production of pro-inflammatory cytokines such as IL-2, IFN-γ, and IL-6. Importantly, neither CD4+ nor CD8+ T-cell subset proliferation and activation post-blinatumomab require exogenous IL-2 administration.
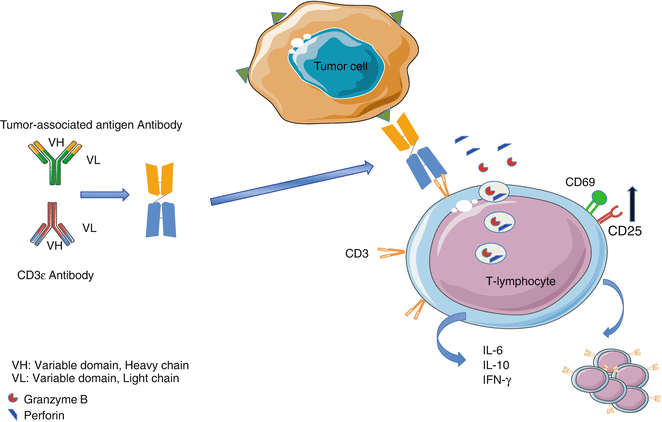
Fig. 38.2
Blinatumomab architecture and mechanism of action
38.3 Blinatumomab: A New Hope in Relapsed/Refractory ALL
Despite the remarkable advances in the treatment of pediatric ALL over the last three decades, standard therapy still fails in 10–20% of the cases. In adult ALL, only half of the patients remain tumor-free at 5 years, and prognosis post-relapse remains grim. B-ALL leukemic cells express several surface antigens amenable to mAb such as CD19, CD20, and CD22. Despite being expressed on normal B-cell lineage from the late pro-B-cell stage to plasma cell differentiation, these receptors are absent on hematopoietic stem cells. Furthermore, we learned from rituximab that CD20 inhibition was associated with only transient B-cell depletion and rare immunosuppressive complications.
The initial BsAbs clinical trial using standard mAb infusion schedule was negative and poorly tolerated. Interestingly, for the clinical development of these molecules, the initial in vivo proof of concept in macaques revealed a unique pharmacokinetics. BsAbs have an extremely short half-life of 2 h, which is in stark contrast to a 21-day half-life of a mAb like rituximab [47]. Based on these preclinical pharmacokinetics data, blinatumomab was subsequently administered in continuous infusion over 28 days using a mini pump to maintain a steady drug concentration.
In adults with relapsed/refractory ALL, two phase II trials led to the FDA approval of blinatumomab in December 2014 and EMA in November 2015. In the MT103-206 trial, 36 patients received continuous blinatumomab and 69% achieved CR or CR with partial hematological recovery [48]. Of note, as reported in prior phase 1 trial, CNS adverse events appeared to be the most relevant complication and required discontinuation of the treatment. These unprecedented results in a heavily pretreated population led to a larger confirmatory phase II trial with 189 patients [49]. CR or CRh occurred in similar proportion of patients without previous (42%) or post (45%)-allogeneic HSCT. It is important to mention that 32 out of the 52 patients who achieved CR or CRh proceeded to allogeneic HSCT.
More recently, the phase 3 TOWER trial confirmed the superiority of blinatumomab to standard care in adult patients with relapsed/refractory ALL. Of the 405 patients included, the median OS was 7.7 months in the blinatumomab group compared to 4.0 months in the chemotherapy group. Treatment with the BiTe also resulted in higher complete remission (34% vs 16% p = 0.01). With respect to toxicity, the investigators reported equal incidence of grade 3 adverse event in both arms [50].
In pediatric relapse/refractory ALL, Arend von Stackelberg published a phase I/II study including more than 90 children. Among the 70 patients who received the optimal dose, 39% achieved complete response, and 52% of them achieved complete minimal residual response. Based on 2-year follow-up, the median OS was 7.5 months and more than 20% were still alive [51].
Safety Profile
Despite the very potent anticancer effect of blinatumomab, unique adverse events have been reported in the majority of clinical trials. From a hematological perspective, thrombocytopenia, neutropenia, and hypogammaglobulinemia were relatively common. Cytokine release syndrome (CRS) was anticipated from the published CAR-T trials. CRS is caused by a transient release of inflammatory cytokines including IL-6 and IL-2 and IFN-γ from T cells after being engaged to tumor cells. Fever, chills, hypotension, and respiratory distress are characteristic. Life-threatening CRS was reported during the first trials of CAR-T, and IL-6 inhibitor tocilizumab was required [52]. To prevent severe CRS, premedication with dexamethasone was recommended especially for patients with high blast count over 15 × 109/L. Neurological events were the most challenging and unexpected toxicity reported for blinatumomab. These included seizure, absence, and confusion. The underlying mechanism is still unclear but might be secondary to a transient neuroinflammatory irritation of the CNS. Activated T cells can cross the blood-brain barrier and trigger toxic inflammation. Guidelines were published in order to adequately prevent and treat neurological toxicity with dexamethasone prophylaxis and dose adjustment.
38.4 BiTe for Non-ALL Hematological Malignancies
The proof of concept on the efficacy of BiTe has been demonstrated in ALL. Similar compounds are being tested for other indications:
- 1.
AMG330 targeting CD3/CD33 is a promising antibody. Preclinical studies demonstrated that targeting CD33 in AML led to T-cell recruitment and expansion [53]. Phase 1 trials are currently being conducted (NCT02520427).
- 2.
Blinatumomab appeared to be effective in 21 heavily pretreated DLBCL patients. After 1 cycle, the overall response rate was 43% and 19 were in CR [54]. More studies are being conducted.
- 3.
Multiple myeloma—multiple epitopes specific to malignant cells are being tested in preclinical studies in phase I. These include BCMA transmembrane activator and calcium modulator exclusively expressed on B-cell lineage or Wue-1 [55].
Blinatumumab are emerging as immunomodulating drugs with significant therapeutic value. However, despite the promising results for patients with ALL relapsed/refractory disease, many patients only experience transient benefit, and the drugs remaine often a bridge to allogeneic HSCT. Clinical trials are already underway to administer BsAbs in less heavily treated patients and in combination with other immunotherapeutic drugs. These approaches might increase the anticancer effects and the cure rate and potentially change the way we treat hematological malignancies.
38.5 Immune Checkpoint Inhibitors (or Activators)
Defective immune response caused by T-cell exhaustion is a common feature of immune escape for many types of cancers and will be extendedly reviewed in this book. The unprecedented positive clinical trials in solid tumors demonstrated the therapeutic potential of blocking the inhibitory receptors CTLA-4 and PD-1. In this section, we will review the most important clinical trials of ICB in hematological malignancies.
Anti-PD-1 Blockade as a Proof of Concept in cHL
cHL was one of the first hematological malignancies in which immune checkpoint blockade was attempted as it represented an interesting model for therapeutic inhibition of the PD-1/PD-L1 axis. Recurrent amplification of the chromosome 9p24.1 locus which includes PD-L1, PD-L2, and JAK2 or infection with EBV all led to hyperexpression of PD-L1 and PD-L2 at the surface of Hodgkin and Reed-Sternberg cells [56]. The pivotal phase 1 trial with nivolumab, a fully human IgG4 directed against PD-1, in 23 patients with relapsed/refractory cHL was a clinical success that led to its approval in 2016 [57] (Table 38.1). With an acceptable safety profile, nivolumab demonstrated impressive clinical activity in heavily pretreated patients, with an objective response rate of 87% (20/23), including 17% with a complete response and 70% with a partial response, and a 24-week PFS rate of 86%. These results have been confirmed in a published phase II trial that enrolled 80 patients after failure of both autologous SCT and BV [58]. The phase Ib KEYNOTE-013 trial with pembrolizumab, another anti-PD-1 mAb, demonstrated similar efficacy though the ORR (65%) was slightly lower than with nivolumab [59]. Nivolumab was tried in cHL after failure of allogeneic SCT with some efficacy despite a risk of triggering acute GVHD [60, 61]. The occurrence in patients who received nivolumab prior to allogeneic SCT of a higher-than-expected rate of early severe transplant-related complications, including several fatal cases of acute GVHD, warrants further scrutiny [62]. To date, PD-1 inhibitors are prescribed continuously for as long as they remain beneficial. In melanoma, some patients were on PD-1 inhibitors for 2 years. The duration of PD-1 treatment in responders remains an open question. Indeed, recent clinical observations suggest that a shorter course or intermittent prescriptions of PD-1 might be as potent as continuous injections. Several investigators are now assessing different PD-1 injection schedule and earlier discontinuation for selected patients.
Table 38.1
Clinical trials of Immune checkpoint inhibitors in hematological malignancies
Study | Patients | Molecule/dosing | Outcome | Toxicity | |
|---|---|---|---|---|---|
Hodgkin lymphoma | Ansell et al. [57] Phase I | 23 patients relapse/refractory cHL (78%—Brentuximab), (78%—autologous HSCT) | Nivolumab 3 mg/kg every 2 weeks
Related posts: Peptide-Based Therapeutic Cancer Vaccines Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment The Human Tumor Microenvironment
 Synergy Between Radiotherapy and Immunotherapy Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|