The availability of anticomplement therapies has been a major achievement for medicine in the last decade. Indeed, eculizumab has changed the treatment paradigm of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome and promises to do the same in several other human complement-mediated diseases. Nowadays, a 10-year experience has also taught us that there are some pitfalls that represent a challenge to improve the current anticomplement treatment. Most of these observations come from paroxysmal nocturnal hemoglobinuria, where unmet clinical needs are emerging, triggering the attention of several investigators and pharmaceutical companies.
Key points
- •
Eculizumab is the current anticomplement treatment agent approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome.
- •
Emerging observations are suggesting that other strategies of complement inhibition/modulation may improve the clinical result of current anticomplement treatment.
- •
Novel complement therapeutics include inhibitors of the terminal effector complement as well as of early complement activation.
- •
Inhibitors of early complement inhibitors include broad C3 inhibitors as well as agents selectively targeting specific complement pathways.
Introduction
The complement system is a key component of innate immunity, which is involved in several physiologic and pathologic processes. It was originally thought that complement merely represents the crudest sentinel for protection from microbes, with a possible additional role in inflammatory processes; however, its role in human homeostasis and disease is now widely recognized. Indeed, dysregulated or impaired complement is involved in an increasing list of human diseases (eg, paroxysmal nocturnal hemoglobinuria [PNH], hemolytic uremic syndrome [HUS], kidney disorders, age-related macular degeneration [AMD]) as well as of clinical conditions (eg, sepsis, ischemia/reperfusion injury, allograft rejection). The interest for complement-mediated pathophysiology has been strengthened by the recent availability of complement inhibitors. Indeed, the clinical approval of the first complement-targeting drug, the anti-complement component 5 (C5) Eculizumab (Soliris), has drastically changed the natural history PNH and represents a novel treatment option for other complement-mediated diseases, such as atypical hemolytic uremic syndrome (aHUS). Subsequently, the introduction of the C1 inhibitor (C1-INH; Cinryze) for the treatment of hereditary angioedema has offered another compound in the armamentarium for the interception of specific component of the complement cascade. All these observations have reignited the interest for a deeper investigation of complement-mediated pathophysiology in human diseases as well as the interest for the development of novel classes of complement inhibitors. Here, current and future agents are reviewed that intercept complement function in vivo and the rationale for targeted complement inhibition for optimizing the therapeutic effect in specific clinical conditions is discussed.
Introduction
The complement system is a key component of innate immunity, which is involved in several physiologic and pathologic processes. It was originally thought that complement merely represents the crudest sentinel for protection from microbes, with a possible additional role in inflammatory processes; however, its role in human homeostasis and disease is now widely recognized. Indeed, dysregulated or impaired complement is involved in an increasing list of human diseases (eg, paroxysmal nocturnal hemoglobinuria [PNH], hemolytic uremic syndrome [HUS], kidney disorders, age-related macular degeneration [AMD]) as well as of clinical conditions (eg, sepsis, ischemia/reperfusion injury, allograft rejection). The interest for complement-mediated pathophysiology has been strengthened by the recent availability of complement inhibitors. Indeed, the clinical approval of the first complement-targeting drug, the anti-complement component 5 (C5) Eculizumab (Soliris), has drastically changed the natural history PNH and represents a novel treatment option for other complement-mediated diseases, such as atypical hemolytic uremic syndrome (aHUS). Subsequently, the introduction of the C1 inhibitor (C1-INH; Cinryze) for the treatment of hereditary angioedema has offered another compound in the armamentarium for the interception of specific component of the complement cascade. All these observations have reignited the interest for a deeper investigation of complement-mediated pathophysiology in human diseases as well as the interest for the development of novel classes of complement inhibitors. Here, current and future agents are reviewed that intercept complement function in vivo and the rationale for targeted complement inhibition for optimizing the therapeutic effect in specific clinical conditions is discussed.
Current complement inhibitors: eculizumab
Eculizumab for the Treatment of Paroxysmal Nocturnal Hemoglobinuria
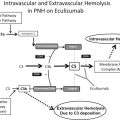
PNH is a rare hematologic disease characterized by complement-mediated intravascular hemolysis, bone marrow failure, and propensity to thromboembolic events. PNH is due to a somatic mutation in the phosphatidyl-inositol glycan class A ( PIG-A ) gene, which impairs the biosynthesis of the glycosyl-phosphatidyl-inositol (GPI) anchor and the subsequent expression of a several surface proteins (GPI-linked proteins). The absence of 2 GPI-anchored complement regulatory proteins (CD55 and C59) is central to the pathophysiology of PNH. CD55 is a regulator of early complement activation, which physiologically inhibits the formation of C3 convertase (both C3bBb and C4b2a) and also promotes its decay. CD59 is a regulator of the terminal effector complement, which interacts with C8 and C9, inhibiting the incorporation of this latter onto the C5b–C8 complex, eventually preventing the assembly of the membrane attack complex (MAC). The concomitant lack of CD55 and CD59 accounts for the susceptibility of PNH erythrocytes to complement activation, which eventually leads to the chronic intravascular hemolysis typical of PNH.
Eculizumab (Soliris) is the first complement inhibitor available in the clinic; it is a humanized monoclonal antibody (mAb), which binds the complement component 5 (C5) and inhibits its cleavage to C5a and C5b, eventually disabling the terminal effector complement (preventing the assembly of the MAC). Eculizumab has been extensively tested in different autoimmune disorders before changing the treatment paradigm of PNH; indeed, 2 large multinational phase III studies demonstrated the efficacy of eculizumab for the treatment of PNH. In the first double-blind, placebo-controlled, multinational randomized trial (TRIUMPH), which enrolled 86 transfusion-dependent PNH patients, treatment with eculizumab resulted in a dramatic reduction of intravascular hemolysis, as measured by lactate dehydrogenase (LDH), leading to hemoglobin stabilization and transfusion independence in about half of the patients (and reduced transfusional need in the remaining ones). These data were confirmed in the open-label phase III study, SHEPHERD, which included a broader PNH population ; this longer study also confirmed the excellent safety profile of eculizumab, with negligible side effects. A subsequent open-label extension study confirmed the efficacy and the safety of eculizumab with a longer follow-up, demonstrating a sustained control of intravascular hemolysis with all related signs and symptoms. This study also demonstrated a remarkable (85%) reduction in the rate of thromboembolic complications, possibly because of the pathogenic linkage between intravascular hemolysis and thrombosis (eg, nitric oxide consumption, prothrombotic microvesicles) or to any direct effect on complement-mediated thrombophilia (ie, on PNH platelets). Preliminary data on survival seem to suggest that eculizumab may also improve survival in PNH, as expected given its effect on the main cause of death in PNH (ie, thrombosis). Based on these data, eculizumab is the standard treatment for PNH, already approved in most countries; according to country-specific guidelines, the indication for treatment includes red blood cell transfusion dependency, and possibly, severe recurrent hemolytic paroxysms and life-threatening thrombotic complications.
Hematologic response to eculizumab in paroxysmal nocturnal hemoglobinuria
Irrespective of a remarkable effect on complement-mediated intravascular hemolysis, the hematologic benefit of PNH patients on eculizumab is extremely heterogeneous: indeed, a significant proportion of patients continue to require some blood transfusions. Different factors may be responsible for this insufficient hematologic response to eculizumab.
- 1.
Bone marrow failure is the most obvious reason; it is clearly identified by low/inadequate reticulocyte counts in patients who do not show any laboratory sign of hemolysis, and it may require additional specific treatments (ie, bone marrow transplantation or immunosuppression).
- 2.
A second possible reason is residual intravascular hemolysis; it may appear as “pharmacokinetic breakthrough,” recurrently appearing in the few hours preceding the next dosing of eculizumab (10%–15% of patients, who may benefit from changes in dosages of administration interval of eculizumab), or as “pharmacodynamic breakthrough,” occurring in concomitance with boosted complement activation (typically at time of infections).
- 3.
A third possible reason is the presence of an intrinsic resistance to eculizumab due to mutated C5; this possibility has been recently demonstrated in a Japanese cohort, but it seems to be restricted to a limited number of patients (and possibly to specific ethnicities).
- 4.
The forth and most common cause of residual anemia during eculizumab treatment is C3-mediated extravascular hemolysis ; it is mechanistically related to the pharmacologic effect of eculizumab and pertains to all PNH patients on eculizumab, even if it becomes clinically relevant in about one-third.
C3-mediated extravascular hemolysis
Inhibition by eculizumab intercepts the complement cascade at the level of C5, eventually counteracting the lack of CD59 on PNH erythrocytes. However, eculizumab does not affect earlier events in complement activation, and especially those that remain uncontrolled because of the lack of CD55. Thus, PNH erythrocytes, which are spared from MAC-mediated hemolysis, progressively accumulate on their surface C3 fragments, which eventually work as opsonins leading to phagocytosis by liver and spleen macrophages. This mechanism of extravascular hemolysis occurs through the recognition of C3 opsonins by specific complement receptors expressed on phagocytic cells, resulting in a selective destruction of PNH erythrocytes, which have been exposed to threshold complement activation. C3-mediated extravascular hemolysis remains subclinical in most PNH patients on eculizumab, but it results in severe anemia in about one-third of them ; recently, it has been reported that a hypofunctional polymorphic variant of complement receptor 1 (CR1) is associated with higher C3 opsonization and lower chance of optimal response to eculizumab. All these findings support a view whereby complement may remain harmful to PNH erythrocytes even during eculizumab treatment, paving the way for the development of novel complement therapeutics.
Eculizumab in Other Indications: Atypical Hemolytic Uremic Syndrome and Cold Agglutinin Disease
Hemolytic uremic syndrome
After its introduction in the clinic, eculizumab was tested in other indications; for the purpose of this review, discussion is limited to hemolytic anemias. Indeed, eculizumab has also been approved for the treatment of HUS, another rare disease that is characterized by thrombotic microangiopathy (TMA), leading to mechanic intravascular hemolytic anemia, platelet consumption, and end-stage renal disease. HUS can be distinct in a sporadic (typical) HUS form, which is acquired and usually associated with infectious events (typically by Shiga-toxin-producing Escherichia coli , STEC), and an atypical form (aHUS), which is inherited. Indeed, aHUS is associated with inherited mutations of different complement genes, including complement factor H (CFH), complement factor I (CFI), complement factor B (CFB), membrane cofactor protein, thrombomodulin, C3 convertase, and C3, clearly pointing out the pathogenic role of complement in this condition. The efficacy of eculizumab in aHUS has been initially demonstrated in 2 distinct prospective trials enrolling aHUS patients who were either resistant to plasma exchange (PE) and/or plasma infusion (PI), or who required PE/PI. In the first trial, which enrolled patients with evidence of platelet consumption, the increase in platelet count was used as efficacy endpoint, as an indicator of ceased TMA. After the initial 26 weeks of treatment, all 17 patients showed some increase in platelet count, with normalization of hematologic values in 88% of patients (15 of 15 who continued to receive the treatment, median treatment duration, 64 weeks). TMA-free status was achieved in 88% of patients (15/17), and only 2 patients required further PE (1 after eculizumab discontinuation due to study withdrawal). More than half of the patients also exhibited a substantial improvement in renal function (as measured by creatinine and GFR), with 4 of 5 patients on dialysis who did not require further procedures. In the second trial, which included 20 aHUS patients on chronic PE/PI, the primary endpoint was TMA-free status (defined as no platelet decrease and no requirement of PE/PI or dialysis), and it was achieved in 16 of 20 (80%) patients (in the remaining 4 patients the primary endpoint failed only because of a mild decrease of platelet count). Similarly to the previous trial, most patients (80%) achieved full normalization of hematologic values (platelets and LDH); however, improvement in renal function took longer and was observed in one-third of patients. These results on renal function are consistent with the mechanism of action of eculizumab, which immediately blocks the pathogenic complement derangement (as demonstrated by hematologic values) but requires longer time to recover from end-organ damages. Based on these studies, eculizumab has been approved by European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) for the treatment of aHUS, and it represents the recommended therapy for this condition irrespective of the demonstration of a causative mutation, provided that other possible diagnoses (eg, thrombotic thrombocytopenic purpura) have been excluded. Nevertheless, a possible role of eculizumab has been suggested even for the treatment of STEC-HUS and of other forms of HUS.
Antibody-mediated hemolytic anemias
It has been hypothesized that eculizumab may be effective even for the treatment of autoimmune hemolytic anemias; so far, objective evidence is scanty and mostly limited to cold agglutinin disease (CAD). CAD is due to autoantibodies of immunoglobulin M (IgM) type, which are specific for some erythrocyte antigens (usually I, more rarely i). This autoimmune disorder may be primary, appearing as a chronic or subacute hemolytic anemia, or even as acute events secondary to some infections (eg, Mycoplasma pneumoniae , mononucleosis ), usually with a self-limiting course. The peculiarity of these auto-antibodies is that they have a typical thermal range, which allows the binding to erythrocytes in the cooler peripheral circulation, and subsequent complement activation in the warmer central circulation (mainly through the classical pathway). This complement activation may lead to MAC assembly and subsequent intravascular hemolysis, but more frequently simply results in an additional opsonization by C3 fragments, which may boost erythrocyte clearance through the reticulo-endothelial system. Indeed, CAD is commonly characterized by extravascular rather than intravascular hemolysis. Eculizumab has proven to be effective in blocking intravascular hemolysis in some cases of CAD, eventually leading to therapeutic benefit. Given the heterogeneous disease presentation and the broad spectrum of effector mechanisms (which may play different roles according to the specificity and the thermal range of patient-specific autoantibodies), it is not surprising that large experiences supporting the use of eculizumab in this condition are lacking.
Complement activation may theoretically occur in all antibody-mediated anemias, and its actual role in disease pathophysiology may differ according to the specific conditions; for instance, MAC-mediated intravascular hemolysis is also typical of isoagglutinin-mediated hemolysis as well as of paroxysmal cold hemoglobinuria. In addition, the complement cascade may contribute even to anemia due to some warm (and mixed warm/cold) autoantibodies, through both MAC-mediated hemolysis and C3-mediated extravascular hemolysis. Indeed, eculizumab has been considered in some refractory, life-threatening, severe hemolytic anemia as off-label, compassionate use ; future studies are needed to better understand the possible role of eculizumab in these specific conditions.
Novel complement inhibitors
Rationale for Developing Novel Anticomplement Agents
The lesson from this first decade of anticomplement treatment with eculizumab is that therapeutic complement inhibition is possible and safe and may result in substantial clinical benefit in several conditions, such as PNH and aHUS. However, this new treatment carries novel pathogenic observations as well as possible pitfalls that may limit the benefit of current treatment. In the case of eculizumab, such pitfalls are not related to the drug itself, but rather mechanistically to its effect on C5. Indeed, C3-mediated extravascular hemolysis is an obvious unmet clinical need that is emerging in PNH (together with the rare genetic resistance to eculizumab), and it paves the way for the development of novel strategies of complement inhibition and/or modulation. Later discussion describes novel complement inhibitors, which are currently in their preclinical or clinical development. Novel complement therapeutics are categorized according to their targets and level of interception in the complement cascade, possibly anticipating their therapeutic effect in candidate clinical indications. The most relevant publically disclosed compounds are listed in Table 1 ; the specific targets and the status of development are included. A poor distinction among complement inhibitors distinguishes between agents that inhibit the effector complement and agents that are able to prevent the arming of early complement activation. C5 and C3 are the key molecules that one may wish to target to inhibit terminal effector complement or early complement activation, respectively. The interception of complement activation at the level of C3 may also exploit inhibition of the specific complement activation pathways, namely, alternative, classical, and mannose pathways.
| Target | Name | Company | Class of Molecule | Status of Development | Ref. |
|---|---|---|---|---|---|
| C5 | LFG316 | Novartis/Morphosys | Monoclonal antibody | Clinical (phase II, AMD) | |
| C5 | Mubodina | Adienne | Monoclonal antibody (minibody) | Preclinical (non-PNH) | |
| C5 | Coversin (OmCI) | Volution Immuno-Pharmaceuticals | Small animal protein (recombinant) | Preclinical (PNH); clinical (phase I, healthy volunteers) | |
| C5 (+C3?) | ATA | NA | Chemical | Preclinical (PNH) | |
| C5 | ARC1005 | Novo Nordisk | Aptamers | Preclinical (non-PNH); clinical (phase I, AMD) | |
| C5 | SOMAmers | SomaLogic | Aptamers (SELEX) | Preclinical (non-PNH) | |
| C5 | SOBI002 | Swedish Orphan Biovitrum (Affibody) | Affibody (fused with albumin-binding domain) | Preclinical (non-PNH); clinical (phase I, healthy volunteers) | |
| C5 | RA101348 | Rapharma | Small molecule (unnatural peptide) | Preclinical (unknown) | (Ricardo A, Arata M, DeMarco S, et al: Development of RA101348, a potent cyclic peptide inhibitor of C5 for complement-mediated diseases. American Society of Hematology meeting 2014. Submitted for publication) |
| C5 | Anti-C5 siRNA | Alnylam | Si-RNA | Preclinical (non-PNH and PNH) | |
| C3 (C3b/iC3b) | H17 | EluSys Therapeutics | Monoclonal antibody | Preclinical (PNH and non-PNH) | |
| C3/C3b | 4(1MeW)/POT-4 | Potentia | Compstatin family | Preclinical (non-PNH); clinical (phase I and II, AMD) | |
| C3/C3b | 4(1MeW)/APL-1, APL-2 | Apellis | Compstatin family | Preclinical (PNH and non-PNH); clinical (PNH and non-PNH, phase I) | |
| C3/C3b | Cp40/AMY-101, PEG-Cp40 | Amyndas | Compstatin family | Preclinical (PNH and non-PNH); clinical (PNH and non-PNH, planned) | |
| CAP C3 convertase | TT30 (CR2/CFH) | Alexion | CFH-based protein | Preclinical (PNH and non-PNH); clinical (phase I, PNH) | |
| CAP C3 convertase | Mini-CFH | Amyndas | CFH-based protein | Preclinical (PNH and non-PNH) | |
| CAP C3 convertase | Mini-CFH | NA | CFH-based protein | Preclinical (non-PNH) | |
| CAP C3 convertase | CRIg/CFH | NA | CFH-based protein | Preclinical | |
| CAP and CP C3 convertase | sCR1 (CDX-1135) | Celldex | CR1-based protein | Preclinical (non-PNH); clinical (phase I, DDD) | |
| CAP and CCP C3 convertase | Mirococept (APT070) | NA | CR1-based protein | Preclinical (non-PNH); clinical (phase I, kidney transplantation) | |
| CAP and CCP C3 convertase | TT32 (CR2/CR1) | Alexion Pharmaceuticals | CR1-based protein | Preclinical (non-PNH) | |
| CFB | TA106 | Alexion Pharmaceuticals | Monoclonal antibody | Preclinical (unknown) | |
| CFD | FCFD4514S | Genentech/Roche | Monoclonal antibody | Preclinical (non-PNH); clinical (phase II, AMD) | |
| CFB | Anti-FB siRNA | Alnylam | Si-RNA | Preclinical (non-PNH) | |
| CFB and CFD | SOMAmers | SomaLogic | Aptamers (SELEX) | Preclinical (non-PNH) | |
| CFB and CFD | NA | Novartis | Small molecules (chemicals) | Preclinical (non-PNH) | |
| Properdin | NA | Novelmed | Monoclonal antibody (and mAb derivatives) | Preclinical (unknown) | |
| C1r/C1s | Cynryze | ViroPharma/Baxter | Human purified protein (C1-INH) | Clinical (approved for HAE) | |
| C1s | TNT003 | True North Therapeutics | Monoclonal antibody | Preclinical (Ab-mediated hemolytic anemias) | |
| MASP-3 | NA | Omeros | NA | Preclinical (PNH and non-PNH) |
Inhibitors of Terminal Effector Complement
The inhibition of the terminal effector complement has been proven safe and effective by the anti-C5 mAb eculizumab. Thus, the interception of complement cascade preventing the cleavage of C5 by the C5 convertase represents an obvious strategy that can be exploited by novel anticomplement agents.
Antibody-based C5 inhibitors
LFG316
Two additional anti-C5 monoclonal antibodies are currently in clinical or preclinical development: LFG316 (Morphosys) is a fully human combinatorial anti-C5 mAb, which is in clinical development for AMD and other ophthalmologic diseases. After initial testing by local administration, the agent is now in phase II as systemic therapy by intravenous injection ( NCT01624636 ).
Mubodina
An additional candidate agent is Mubodina, an anti-C5 “minibody,” consisting of an engineered antibody fragment including only the antigen-specific variable light (VL) and heavy (VH) chain domains of its parental anti-C5 mAb. The minibody Mubodina prevents the cleavage of C5 and inhibits the generation of inflammatory molecule C5a and of C5b, and the subsequent lytic MAC. Mubodina has been approved as an orphan drug for some kidney diseases by both the FDA and EMA, and its pharmacologic inhibition of C5 cleavage (and thus of generation of C5a and C5b, and subsequently of MAC assembly ) anticipates an effective complement inhibition in vivo. Further derivatives of the original anti-C5 scFv are currently under development, investigating the possibility of targeting the anti-C5 agent at specific sites; this strategy, which exploits anti-C5 scFv fused with specific tag peptides, has proven effective in an animal model of arthritis and might be translated in clinical application in the near future. These antibody-based inhibitors of C5 seem a ready-to-go therapeutic alternative for the rare PNH patients harboring C5 mutations, resulting in intrinsic resistance to eculizumab, because the specific mutation occurs in the C5 epitope recognized by eculizumab and does not affect the binding to other antibodies.
Coversin
Another novel candidate complement inhibitor is coversin (also known as OmCI), a small (16 kDa) protein of the lipocalin family isolated from the tick Ornithodoros moubata . Coversin binds to human C5 and prevents its cleavage by C5 convertases ; it also exerts a broader anti-inflammatory effect by binding to leukotriene B4. Preliminary in vitro data in PNH suggest possible inhibitory effects on intravascular hemolysis as well as some effect in preventing C3 opsonization (this latter effect is not entirely clear, because it is not expected for a compound acting downstream in the complement cascade at the level of C5). However, coversin has shown amenable pharmacokinetic and pharmacodynamic profiles in a phase I study in healthy volunteers following subcutaneous administration; a clinical translation plan for PNH patients is planned.
Next-generation targeted C5 inhibitors
The spectrum of C5 inhibitors also includes several novel classes of compounds exploiting new technologies for specific design of more specific targeted therapeutics.
Aptamers
Aptamers are large (either oligonucleic or peptide) compounds that selectively bind to specific molecules, eventually impairing their function. They are usually created by using large random sequence pools, from where target-specific molecules are selected. ARC 1905 (Zimura) is a PEGylated, stabilized oligonucleic aptamer targeting C5, which is currently in phase II clinical investigation for ophthalmologic disease (AMD).
SOMAmers
A further evolution of aptamers exploits a systematic evolution of ligands by exponential enrichment (SELEX); SOMAmers (slow off-rate modified aptamers) are these novel class of compounds with a more favorable pharmacokinetic (PK) and pharmacodynamic (PD) profile. SOMAmers specific for different key components of the complement cascade (C5, C3, complement factor D [CFD], and complement factor B [CFB]) are currently under preclinical development by SomaLogic and can be considered for therapeutic application.
Affibodies
A similar technology exploiting target-specific inhibition is developed by the Swedish company, Affibody, who is developing small antibody mimetic proteins (about 6 kDa) by a combinatorial protein engineering approach. The resulting small nonimmunoglobulin proteins display a high-affinity binding to a wide range of protein targets, including C5. More recently, Swedish Orphan Biovitrum created a C5-specific affibody fused to an albumin-binding domain (SOBI002; 12 kDa); SOBI002 binds human C5 with low-nanomolar affinity (dissociation constant [KD] ∼1 nM), eventually preventing its cleavage and thus the activation of the downstream effector complement. The compound demonstrated excellent bioavailability in nonhuman primates (NHP), and the fusion with the albumin-binding moiety increases half-life and stability, resulting in a terminal half-life greater than 2 weeks. SOBI002 has been shown effective in preclinical models of complement (C5)-mediated diseases, and a phase I study evaluating safety, tolerability, and PK/PD in healthy volunteers has recently started ( NCT02083666 ).
Cyclomimetics
Rapharma is developing another class of compounds, which are small, cyclic, peptidelike polymers with backbone and side-chain modifications, called Cyclomimetics. Cyclomimetics, which are produced by ribosomal synthesis of unnatural peptides, have beneficial properties as compared with natural peptides, including a low risk of immunogenicity (due to poor major histocompatibility complex (MHC) presentation) and increased cell permeability, stability, potency, and bioavailability (due to the structural modifications). RA101348 is the lead anti-C5 macrocyclic peptide that is currently in preclinical investigation (Ricardo A, Arata M, DeMarco S, et al: Development of RA101348, a potent cyclic peptide inhibitor of C5 for complement-mediated diseases. American Society of Hematology meeting, 2014. Submitted for publication).
siRNAs
The spectrum of novel anti-C5 agents is not limited to proteins or peptides; indeed, Alnylam is developing RNA therapeutics for clinical complement inhibition, targeting liver-expressed genes such as C5. Robust (>95%) silencing of C5 liver production has been achieved after subcutaneous injections of GalNAc-conjugated anti-C5 siRNA duplex; in animal models, this results in sustained and durable (recovery started 2 weeks after single-dose injection) complement inhibition as effective as 90%. This agent is currently under preclinical investigations in NHP as well as in PNH in vitro. Even if siRNA-mediated complement inhibition is an elegant and promising novel therapeutic approach, further studies are needed to confirm its clinical feasibility (and efficacy, given that residual low activity might be problematic).
Chemicals
The targeted design of complement therapeutics using these novel technologies offers a broad spectrum of anti-C5 inhibitors with a highly specific inhibition; thus, they seem superior to other chemicals that may intercept C5 or other complement components. For instance, aurin tricarboxylic acid (ATA) has been reported effective in preventing hemolysis of PNH erythrocytes in vitro, with also a possible unexplained effect on the C3 convertase; however, it is likely that these effects of ATA result from a broad inhibition of proteases, which may impair the function of several unrelated proteins, not restricted to complement components. Nevertheless, the rational design of novel modified chemicals with specific effect on C5 or other components of the complement cascade is possible and currently under investigation.
Inhibitors of Early Complement Activation
The attempt to inhibit complement activation at the level of C3 is a rational strategy to improve the efficacy of current anticomplement treatment, especially in PNH. The key event in early complement activation is C3 cleavage by C3 convertases generated along one of the classical complement activating pathway (CCP), alternative (CAP), and mannose (CMP). Inhibitors of early complement activation may directly target C3 or may also prevent C3 activation, exerting their effect upstream, on pathway-specific events that eventually lead to C3 activation ( Fig. 1 ; see Table 1 ).
Related posts:
 Complement in Health and Disease
Complement in Health and Disease
 Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
 Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal Nocturnal Hemoglobinuria
 Hemolysis from ABO Incompatibility
Warm Autoimmune Hemolytic Anemia
Thrombotic Microangiopathy
Hemolysis from ABO Incompatibility
Warm Autoimmune Hemolytic Anemia
Thrombotic Microangiopathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


