Fig. 7.1
Locations of missense PAX8 mutations (secondary structure). Amino acid sequences of human PAX genes (PAX1 to PAX9) are shown. The sequences correspond to the paired domain, which consists of two β sheets (β1 and β2) and six α helices (α1 to α6) (shown in boxes). Although the roles of PAX genes in development and physiology are distinct, the protein sequence are highly conserved among the gene family members. To date, thirteen PAX8 mutations affecting eleven amino acid residues have been reported (shown in red letters). The majority of the mutations are located in α1 to α3 helices. Only two mutations have been observed in two or more unrelated families (p.R31H and p.R133Q; asterisks)
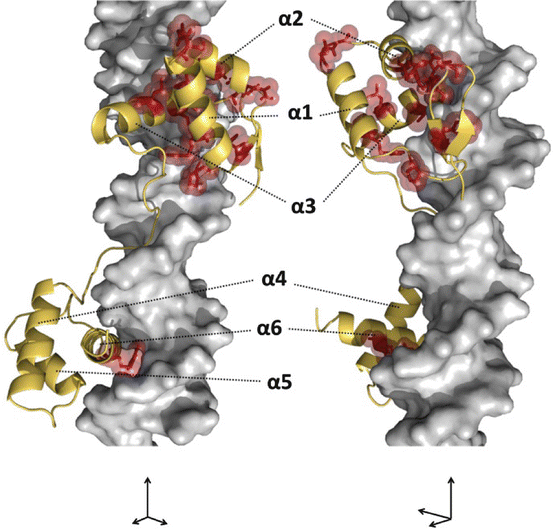
Fig. 7.2
Locations of missense PAX8 mutations (three-dimensional structure). An image of three-dimensional structure of the PAX-DNA complex (PAX, gold; DNA, silver). The image was produced with PyMOL (www.pymol.org) based on the data of PAX6-DNA complex (protein data bank accession 6PAX; http://www.rcsb.org/). Side chains of the amino acid residues that are affected by human PAX8 mutations are shown in red. As shown in Figs. 7.1 (secondary structure) and 2 (three-dimensional structure), most of human PAX8 mutations are located in α1 to α3 helices
Murine PAX8 was first identified from mouse cDNA library in 1990 [1]. PAX8 is exclusively expressed in the thyroid from the time of specification of thyroid anlage to adulthood [1], suggesting its roles in both thyroid development and physiology. Expression of PAX8 is also observed in the kidney, where PAX2 (a closely related paralogue of PAX8) is expressed. PAX8 is essential for the activation of the thyroperoxidase (TPO) gene, thyroglobulin gene, and sodium/iodine symporter (NIS) gene in vitro [2, 3]. In a rat thyroid cell line model, loss of PAX8 expression, which is triggered by transformation with polyoma middle T-antigen, causes remarkable loss of thyroid-specific proteins such as thyroglobulin, TPO, and NIS [4]. These changes can be reversed by reinduction of PAX8.
In 1998, establishment and characterization of genetically engineered PAX8-deficient mice was reported (the paper was published “back to back” in Nature Genetics with reports of first human PAX8 mutations)[5]. Pax8 knockout mice are hypothyroid and die soon after weaning if not replaced with thyroid hormones. In the mice, thyroid diverticulum develops, but the cell population cannot expand. This proliferation defect in early organogenesis results in severe thyroid hypoplasia. Development of calcitonin-producing tissues is not affected. The phenotype of Pax8 knockout mice indicates that proliferation of thyroid precursor cells require at least one copy of PAX8, but specification of thyroid anlage does not in mice. This is why PAX8 is not regarded as the “master switch” of thyroid development. The situation is contrasting to PAX6, which acts as a master switch of eye development [6].
More recently, new insights into the role of PAX8 in early thyroid development were provided from a series of cell engineering experiments. Antonica and colleagues, who established the protocol to induce thyroid follicular cells from mouse embryonic stem cells (ESCs), reported that transient expressions of PAX8 and NKX2-1 (also known as thyroid transcription factor-1) were sufficient to induce thyroid-specific molecules including thyroglobulin, TPO, NIS, and TSH receptor [7]. In the paper, transient expression of “PAX8 alone” or “NKX2-1 alone” was also tested as control experiments. Surprisingly, overexpression of PAX8 in mouse ESCs results in virtually no effect on abovementioned thyroid-specific molecules. Contrastingly, overexpression of NKX2-1 alone can induce those genes, although the mRNA expression levels were low as compared with PAX8/NKX2-1 coexpression. NKX2-1 is the most potent “molecular switch” of thyroid development known to date, but gene expression sufficient to initiate thyroid hormone production in ESC-derived cells requires simultaneous expression of PAX8 and NKX2-1. These observations agree well with previous in vitro observations that PAX8 and NKX2-1 synergistically transactivate thyroid-specific genes [8, 9]. NKX2-1 exhibits wider spatial expression pattern (e.g., lung and brain) than PAX8, but thyroid-specific molecules are not expressed in these extrathyroidal tissues. The restricted expression patterns of NKX2-1 responsive thyroid-specific genes (e.g., thyroglobulin, TPO, NIS, and TSHR) are likely defied by coexpressed transcriptional partner, PAX8.
7.3 Congenital Hypothyroidism due to PAX8 Mutations in Humans
In 1998, Macchia and colleagues screened PAX8 mutations in 145 Italian patients with thyroid dysgenesis and identified two heterozygous PAX8 mutation carriers (p.R31H and p.R108X) [10]. These two patients were sporadic, and family analysis showed that the mutation occurred de novo. Macchia et al. also identified a dominantly inherited mutation (p.L62R) in a German family with autosomal dominant hypothyroidism (two children and their mother were affected).
After the first identification of PAX8 mutation-carrying patients, more than 50 mutation carriers belonging to 23 families have been described in the literature [11–26]. These publications have revealed variable phenotypes of mutation carriers regarding thyroid morphology and hormone-producing capacity. About 80 % of patients that evaluated their thyroid morphology had thyroid hypoplasia. Therefore, thyroid hypoplasia is a typical radiological finding of PAX8 mutation carriers. Only one patient with thyroid ectopy was reported [10], while no patients had thyroid aplasia. Though the majority of mutation carriers have a hypoplastic thyroid, thyroid morphology can be completely normal and can be variable even within a family [14, 16]. Decreased echogenicity of the thyroid has been described in several mutation carriers [24, 25]. This change is presumably due to the altered tissue architecture of mutation-carrying thyroid (described below).
As for thyroid functions, most mutation-carrying patients have severe permanent congenital hypothyroidism, with decreased serum-free thyroxine levels. Although patients are usually diagnosed in the frame of newborn screening for CH, three cases that had a negative newborn screen result have been described [17, 24]. These observations suggest that a minor subset of PAX8 mutation carriers have delayed-onset CH.
Although extrathyroidal manifestation is rare among PAX8 mutation carriers, at least four mutation carriers with congenital anomalies of the kidney and urinary tract (e.g., unilateral kidney agenesis, congenital ureterocele, horseshoe kidney) have been reported [23]. Considering the expression of PAX8 in the developing kidney, these anomalies could be associated with the mutation.
7.4 Frequency of Human PAX8 Mutations
As described above, PAX8 was first identified as a protein that is predominantly expressed in the developing thyroid of mice. The remarkable phenotype (severe thyroid hypoplasia) of the Pax8 knockout mice also confirmed the important role of PAX8 in thyroid development. Therefore, early human mutation screening studies of PAX8 have focused on CH with thyroid dysgenesis (thyroid aplasia, ectopia, or hypoplasia) [10, 13, 14]. In these early studies, PAX8 mutations were found in 1.4–3.3 % of thyroid dysgenic CH subjects, confirming that PAX8 is also involved in development of the human thyroid (Table 7.1). However, considering the wide clinical variability of human PAX8 mutation carriers, these frequency data could have a potential bias resulting in either an underestimate or an overestimate of the true frequency. More recently, mutation screening has been extended to CH patients lacking radiological evidence of thyroid dysgenesis, finding that 0.7–2.0 % of total CH patients (with or without thyroid dysgenesis) have PAX8 mutations [17, 21, 22, 25].
Table 7.1
Summary of the results of PAX8 mutation screening
Author | Year | Study site | Study subjects | Frequency of mutations | Ref. |
|---|---|---|---|---|---|
Macchia et al. | 1998 | Italy | Thyroid dysgenesis | 2 in 145 (1.4 %) | [10] |
Vilain et al. | 2001 | Belgium | Thyroid dysgenesis | 2 in 61 (3.3 %) | [13] |
De Sanctis et al. | 2004 | Italy | Thyroid dysgenesis | 1 in 54 (1.9 %) | [14] |
Al Taji et al. | 2007 | Czech | Congenital or early-onset hypothyroidism (Thyroid dysgenesis, 65 %) | 3 in 170 (1.8 %) | [17] |
Narumi et al. | 2010 | Japan | Congenital hypothyroidism (Thyroid dysgenesis, 51 %) | 2 in 103 (2.0 %) | [25] |
Alcántara-Ortigoza et al. | 2012 | Mexico | Congenital hypothyroidism
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|