The severe clinical symptoms of inherited CD59 deficiency confirm the importance of CD59 as essential complement regulatory protein for protection of cells against complement attack, in particular protection of hematopoietic cells and human neuronal tissue. Targeted complement inhibition might become a treatment option as suggested by a case report. The easy diagnostic approach by flow cytometry and the advent of a new treatment option should increase the awareness of this rare differential diagnosis and lead to further studies on their pathophysiology.
Key points
- •
Congenital isolated deficiency of CD59, a key regulator of the complement system, is a rare disorder.
- •
Congenital isolated deficiency of CD59 is associated with Coombs-negative hemolysis and peripheral polyneuropathy.
- •
Flow cytometric analysis of expression of CD59 and other glycosylphosphatidylinositol-anchored proteins is an important step in diagnostic workup if CD59 deficiency is suspected.
- •
Targeted complement inhibition might become a new treatment option for congenital isolated deficiency of CD59.
Introduction
Homologous restriction factor (HRF 20), membrane inhibitor of reactive lysis (MIRL), membrane attack complex inhibitory factor (MACIF), protectin, and CD59 are a few of the many names for the same protein that was discovered when lysis of erythrocytes by complement in a homologous test system was studied. CD59 is a complement regulatory protein with a length of 128 amino acids that is attached to the cell surface by a glycosylphosphatidylinositol (GPI) anchor. GPI-anchored proteins exert multiple functions; some are important regulators of the complement system. The model disorder for a complement-mediated hemolytic anemia, paroxysmal nocturnal hemoglobinuria (PNH), is caused by a deficiency of all GPI-anchored proteins (including CD59) on the affected cells. PNH is caused by acquired PIGA gene mutations in hematopoietic stem cells, which cause a defect of the biosynthesis of the GPI-anchor. Recently, a case of PNH caused by a germline splice site mutation of PIGT and an acquired deletion of PIGT in hematopoietic cells was reported. This finding shows that genes that are essential for GPI biosynthesis and anchoring other than PIGA might also cause PNH. The frequency of these non- PIGA PNH cases still needs to be determined.
In contrast to the acquired PNH, in which only hematopoietic cells harbor the PIGA mutation, in the rare inherited GPI-deficiency syndromes, all cells are affected. So far, underlying germline mutations were identified in PIGL , PIGM , PIGN , PIGO , PIGT , PIGV , and even CD59. Additionally a germline PIGA gene mutation was recently reported in several families. In PNH and the inherited defects of GPI anchor synthesis, the expression of the whole class of GPI-anchored proteins, including CD59, on the cell surface is affected. In contrast, isolated deficiency of CD59 is caused by inherited mutations in the CD59 gene. The biosynthesis of the GPI anchor itself and the expression of the other GPI-anchored proteins are normal. This disorder is rare. Only 7 cases have been published so far. Nevertheless, it gives important insight to the pathophysiologic role of CD59. Here, we review the genetic basis, differential diagnosis, clinical findings, and new treatment options for isolated inherited CD59 deficiency.
Function of CD59
CD59 is a glycoprotein of approximately 20 kDa. It is attached by a GPI anchor to the membrane of many different cell types, including hematopoietic cells, endothelial cells, neurons, Schwann cells, oligodendrocytes, and astrocytes. CD59 occurs at low concentration in soluble form in plasma, urine, and other body secretions. CD59 is a key regulator of the complement system and prevents complement attack by the inhibition of the membrane attack complex (MAC). The MAC triggers cell activation, endothelial damage, and cytotoxicity and leads to neurodegeneration and to cell lysis by the formation of a transmembrane pore ( Fig. 1 ).
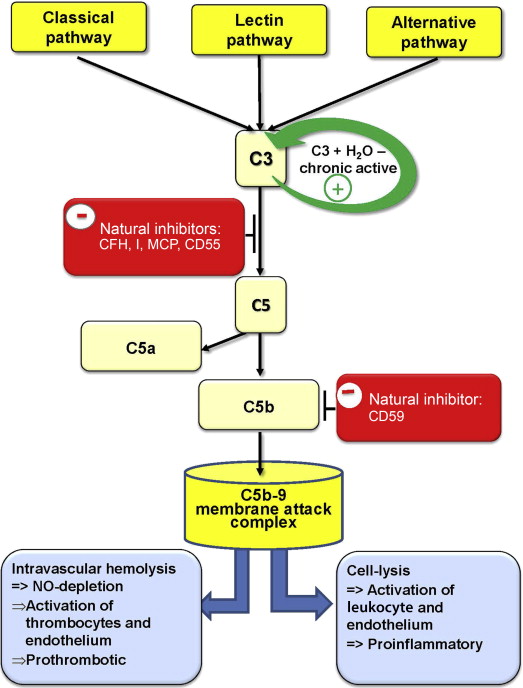
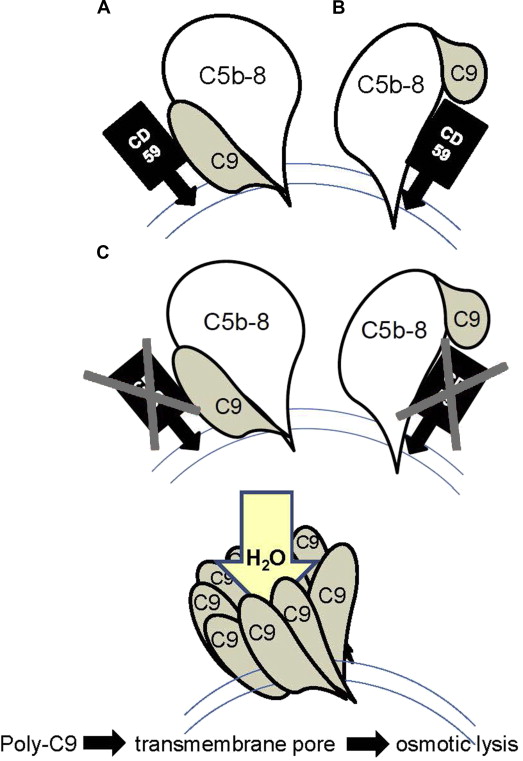
This pore is built by a complex of complement components C5b, C6, C7, and C8 (C5b-8) completed by a poly C9 complex consisting of 12 to 16 polymerized C9 molecules (poly C9). This poly C9 can form tubular structures and can be inserted in phospholipid bilayers. However, poly C9 by itself is not cytolytic without a fully assembled MAC that is capable of forming channels through lipid bilayers.
CD59 is the inhibitor of this reaction by binding to C9 or C5b-8, which reduces the development of poly C9. Therefore, a loss of CD59 increases the amount of poly C9 and consequently of the C5b-9 complexes, which leads to more functional transmembrane pores, resulting in an increase of osmotic cell lysis ( Fig. 2 ).
 , Target protein expression in the sample;
, Target protein expression in the sample;  , isotype control.
, isotype control. Results of several studies point to the role of MAC and CD59 in causing demyelination and neuronal death in neurodegeneration. A CD59 deficit seems to be associated with the neuritic losses characteristic of Alzheimer’s disease and enhances disease severity, demyelination, and axonal injury in allergic encephalomyelitis. Experimental data show that MAC formation provokes seizures and neurodegeneration. In a mouse model of neuropathy, MAC deposits on damaged nerve terminal axons and surrounding perisynaptic Schwann cells were found. Furthermore, damage is exacerbated in tissues from mice lacking CD59, in which MAC formation is increased. CD59a knockout models in mice suggest that CD59a protects against ischemic brain damage and mediates protection from secondary neuronal death after traumatic brain injury.
CD59a and CD59b double-knockout in mice results in complement-mediated hemolysis and hemoglobinuria, which can be rescued by deficiency of C3.
Genetic basis of CD59 deficiency
So far, 3 different CD59 null alleles have been described. One patient from Japan carried the 2 single-base deletions c.123delC and c.361delG leading to a frameshift mutation with a stop codon at amino acid 38 (p.Val42Serfs*38; p.Ala121Glnfs). Five CD59-deficient children originating from 4 unrelated North-African Jewish families all harbored the nucleotide substitution c.266G> A (p.Cys89Tyr), which likely is a founder mutation in Jews of North African ancestry. The authors recently reported an isolated complete CD59 defect caused by the homozygous deletion c.146delA leading to a terminating codon at amino acid 31(p.Asp49Valfs*31) in a patient originating from Turkey. The Japanese and the Turkish patient probably generate no functional protein. The altered protein of the North African patients was present in cells but absent on membranes. It is speculated that the changed tertiary structure may prevent the presentation of the altered protein on the membrane.
Clinical symptoms of CD59 deficiency
Except the single Japanese patient who was affected by the first hemolytic episode not before the age of 13, all 6 patients from North African and Turkish origin showed an early onset of the disease with 3 to 7 months. All published CD59-deficient individuals were severely ill and presented with recurrent hemolytic crises ( Table 1 ). Furthermore, a part of the complications observed in congenital CD59 deficiencies, like acute renal failure and thromboembolic events, are probably mediated by intravascular hemolysis. Reduction of availability of nitric oxide (NO) is a well-known mechanism in intravascular hemolysis, especially in PNH. This reduction is caused by a disturbed synthesis and an increased consumption of NO. Reduced bioavailability of NO results in platelet activation, endothelial dysfunction, and contraction of the vessels. The Japanese patient suffered from 2 cerebral infarctions. In the Turkish child, diffusion disturbances suggestive of an ischemic stroke could be detected by cerebral MRI during the second hemolytic crisis. Thus, similar to acquired PNH, in inherited CD59 deficiency, the risk of thromboembolic complications might be increased. Symptoms involving the peripheral nervous system were reported in all published cases except the Japanese patient published by Yamashina and colleagues (see Table 1 ). In most cases, the neurologic symptoms are more prominent than the hemolysis, which only becomes clinically relevant during episodes of complement activation, especially during infections. The mechanism behind this observation seems to be the limited neuronal capacity of controlling complement activation because of low neuronal CD59 expression.
| Congenital CD59 Deficiency | |||
|---|---|---|---|
| Ethnic Origin | Japanese | Turkish | North African Jewish |
| Number of patients | 1, parents consanguineous | 1, parents consanguineous | 5; from 4 parents consanguineous |
| Location of mutation | Frameshift mutation leading to terminating codon at amino acid 38 (p.Val42Serfs*38; p.Ala121Glnfs) | Frameshift mutation leading to terminating codon at amino acid 31 (c.146delA, pAsp49Valfs*31) | Homozygous missense mutation leading to amino acid substitution from Cys to Tyr in position 89 (p.Cys89Tyr) |
| Onset of disease | 13 y | 7 mo | 3–7 mo |
| Hemolysis | Yes | Yes | Yes |
| Thrombo-embolic events | Yes, cerebral infarction | MRI suggestive of ischemic stroke during the second hemolytic crisis | No |
| Neurologic disease | Nothing reported (beside the cerebral infarctions) | Progressive neurologic impairment: focal seizures, bulbar symptoms, abducens and facial nerve palsy; generalized muscular hypotonia (legs flaccid and muscle reflexes absent), ventilation required | Relapsing polyneuropathy, presenting as chronic inflammatory demyelinating polyradiculoneuropathy with symmetric muscle weakness, accompanied by hypotonia and absent tendon reflexes involving the legs > arms |
| Treatment | Nothing reported | Immunoglobulins intravenously; plasmapheresis, transfusions without response; Eculizumab, with treatment response of hemolysis and neurologic symptoms | Immunoglobulins intravenously, plasmapheresis, corticosteroid, rituximab, and cyclosporine did not prevent recurrences but seemed to have effect on relapses length and severity |
| Reference | Yamashina et al, 1990 Motoyama et al, 1992 | Höchsmann et al, 2014 | Nevo et al, 2013 |
The common feature in these 6 individuals with a congenital CD59 deficiency and neurologic symptoms is a generalized progressive muscular hypotonia with flaccid paralysis, presenting like a chronic inflammatory demyelinating polyradiculoneuropathy. One patient showed, in addition, symptoms of the central nerve system (bulbar symptoms, ie, inability to swallow, abducens, and facial nerve palsy) and focal seizures.
The reason for differences in clinical presentation is currently unknown, and more cases may be necessary to draw definite conclusions. A comparison of the clinical characteristics of the cases is listed in Table 1 .
Laboratory diagnosis of CD59 deficiency
Acquired CD59 deficiency caused by a GPI-anchor defect on hematopoietic cells is a hallmark of PNH. Varying percentages of hematopoietic cells are affected, and PNH is characterized by a mosaic of cells with normal and absent or reduced expression of GPI-anchored proteins.
In contrast to PNH, which affect the synthesis of the GPI anchor and, therefore, the expression of all GPI-anchored proteins on the cell surfaces, only CD59 expression is missing in congenital CD59 deficiency, and the defect is not confined to hematopoietic cells.
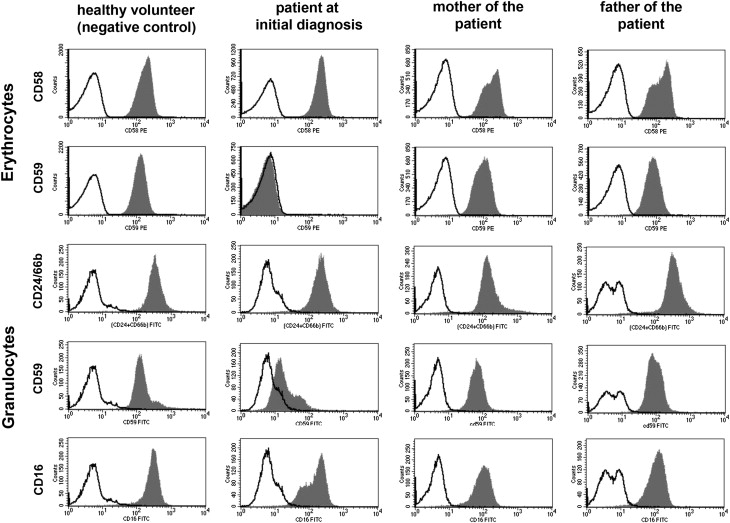
Flow cytometric screening for PNH is mandatory in workup of hemolytic anemia with a negative Coombs test result (direct antiglobulin test). The screening panel should consist of a minimum of 2 different GPI-anchored markers (including CD59) on a minimum of 2 different cell lines. Screening with a single marker (eg, based on CD55 or CD16 alone) would have missed the diagnosis in these cases of isolated CD59 deficiency ( Fig. 3 ). On the other hand, screening with CD59 as a single marker could have suggested the wrong diagnosis of acquired PNH.
Related posts:
 Complement in Health and Disease
Complement in Health and Disease
 Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
 Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


