The complement system is an essential component of the immune system. It is a highly integrative system and has a number of functions, including host defense, removal of injured cells and debris, modulation of metabolic and regenerative processes, and regulation of adaptive immunity. Complement is activated via different pathways and it is regulated tightly by several mechanisms to prevent host injury. Imbalance between complement activation and regulation can manifest in disease and injury to self. This article provides an outline of complement activation pathways, regulatory mechanisms, and normal physiologic functions of the system.
Key points
- •
The complement system is composed of over 50 interacting serum and membrane-bound proteins that provides an effective immune surveillance.
- •
Complement can be activated via 3 different pathways: the classical, lectin, and alternative pathways, which converge at the cleavage and activation of C3 with the subsequent generation of various biological effector molecules.
- •
Strict regulation of the complement system is mediated by a number of soluble and membrane-bound proteins to prevent damage to self.
- •
Complement plays a key role in a number of biological processes, including host defense, removal of injured cells and debris, modulation of metabolic and regenerative processes, and the regulation of adaptive immunity.
- •
Inappropriate complement activation and impaired regulation can lead to self-directed attack and contributes to various diseases and disease-related conditions.
The complement system is a major component of the innate immune system and it provides a powerful and effective mechanism to protect the host from pathogens. It was first described in the late 19th century as a heat-labile component of serum that “complemented” the effects of antibodies in the lysis of bacteria and red blood cells. The term complement was coined by Paul Ehrlich in 1899. We now know that the complement system is made up of some 50 serum and membrane proteins with tightly regulated proteolytic activation cascades that culminate in the production of effector molecules with multiple biological functions. It has long been known that complement provides host surveillance and protection from microbes, and it is now clear that complement also plays important and diverse roles in several other physiologic and homeostatic functions, such as the clearance of dead and dying cells, developmental and regenerative processes, and the modulation of humoral and cell-mediated immune responses. Furthermore, disruption of the balance between complement activation and complement regulation is involved in the pathogenesis of several diseases and disease states, ranging from traumatic injury and ischemia-related conditions, to autoimmune disease, to alloreactivity and transplant rejection. Complement is also implicated in tumor immune surveillance, and recently tumor-promoting functions of complement have also been described.
Soluble complement proteins are synthesized primarily by hepatocytes, although significant amounts are also synthesized by monocytes, macrophages, and some epithelial cells in the gastrointestinal and urinary tracts. Activation of complement is normally achieved via 3 different pathways: the classical, alternative, and lectin pathways. Each of these pathways is initiated by different stimuli ( Table 1 ), but all lead to the cleavage and activation of the central complement protein C3, with the subsequent cleavage of C5 and generation of biological effector molecules. In addition to the 3 well-defined pathways of activation, there are also bypass mechanisms of activation, such as the direct proteolytic cleavage of C5.
| Pathway | Trigger for Activation |
|---|---|
| Classical | Antibodies bound to bacteria, fungi, viruses, or tumor cells Immune complexes Apoptotic cells C-reactive protein Activated Factor XII |
| Alternative | Continuous hydrolysis of complement protein C3 |
| Lectin | Microbes with terminal mannose groups |
| Other activators of complement | Thrombin Kallikrein |
Activation of the complement system
The Classical Pathway
The classical pathway is triggered by antibody-antigen immune complexes via C1q recognition of Fc domains in conformationally altered immunoglobulin (Ig)M or clustered IgG ( Fig. 1 ). The interaction of C1q with Fc causes a conformational change within the C1q molecule and the subsequent cleavage and activation of the associated C1r and C1s serine proteases. Activated C1s then cleaves C4 and C2 into 2 large active fragments (C4b and C2a) and 2 small soluble inactive fragments (C4a and C2b). Cleavage of C4 exposes a reactive thioester within the C4b fragment, which results in covalent attachment of C4b to the activating surface. The binding of C2 to C4b and the subsequent cleavage of C2 result in the covalently attached classical pathway C3 convertase, C4bC2a (note that there is discrepancy in the literature in the designation of C2a vs C2b). This complex cleaves C3 into C3b (large) and C3a (small). Similar to C4b, C3b contains a reactive thioester that can become bound covalently to the activating surface, and that can initiate activation of the alternative pathway. If C3b binds to the C4bC2a complex, it forms to classical pathway C5 convertase (C4bC2aC3b) that cleaves C5 into C5b and C5a, with initiation of the terminal complement pathway. Both C3a and C5a are known as anaphylatoxins and are powerful biological effector molecules with diverse functions, including inflammation, modulation of adaptive immunity, and repair and regenerative processes.
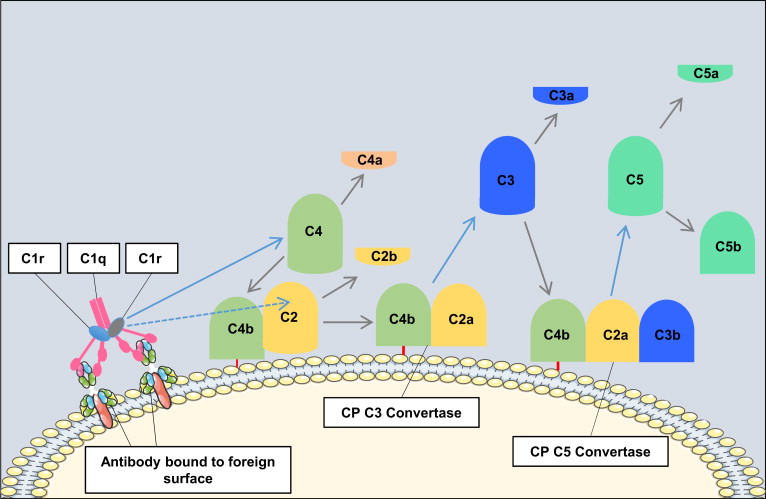
Not all antibody isotypes bind C1q and activate complement. IgM is very effective at activating complement, but some subclasses of IgG are poor activators (human IgG4 and mouse IgG1). Activation of the classical pathway can also occur independent of antibodies, and C1q can bind directly to certain microbial epitopes or epitopes exposed on apoptotic and necrotic cells. C1q can also bind to cell surfaces via pattern recognition molecules such as C-reactive protein, an acute phase protein that binds to the surface of pathogens or injured cells and activates the classical pathway. This process tags surfaces with complement proteins to facilitate their removal by phagocytes. An interaction can also occur between the coagulation cascade and the complement system, as activated factor XII is able to interact with C1 and activate the classical pathway.
The Lectin Pathway
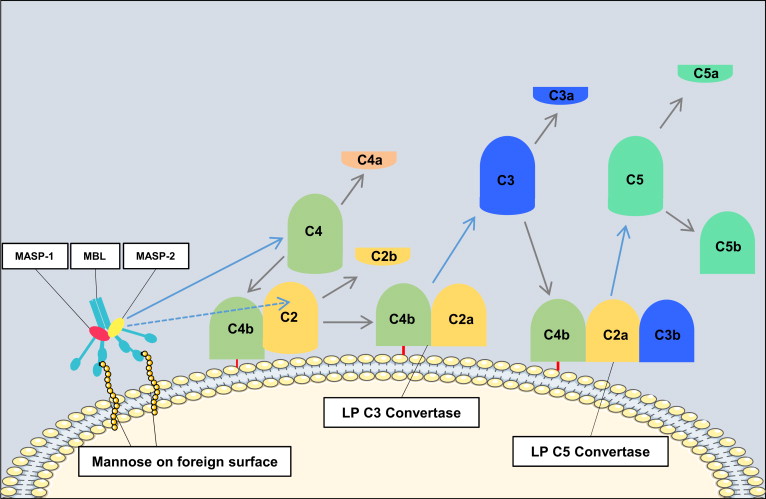
Activation of the lectin pathway results in formation of the same C3 convertase as is generated in the classical pathway (C4bC2a). The catalytic activation sequences of the lectin pathway resemble those of the classical pathway, but with the utilization of different recognition molecules and different associated serine proteases ( Fig. 2 ). In the lectin pathway, mannose-binding lectin (MBL) or ficolins recognize patterns of carbohydrates such as N -acetylglucosamine or mannose, but not sialic acid or galactose, which provides selectivity for bacterial, viral, fungal, and parasitic cell surfaces. MBL is structurally and functionally similar to C1, and the binding of MBL (or ficolin) to a carbohydrate ligand activates MBL-associated serine proteases (MASP-1, MASP-2, and MASP-3), which generate C4bC2a in a manner analogous to that of the C1 complex.
The Alternative Pathway
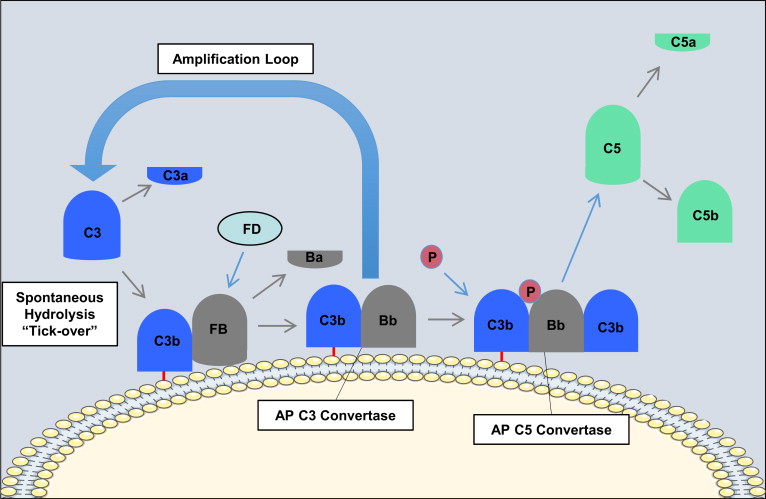
In contrast with the classical pathway and lectin pathway, the alternative pathway is constitutively active and depends on the slow spontaneous hydrolysis of C3, which exposes a binding site for the alternative pathway protein, factor B, and subsequent generation of the alternative pathway C3 convertase (C3bBb; Fig. 3 ). This “tickover” process provides a primed and rapid response mechanism for the deposition of complement on foreign surfaces. Activation and amplification of the alternative pathway depend on the surface to which hydrolyzed C3 becomes attached covalently. Activating surfaces, such as the surface of a microbe, do not have the means to regulate further C3 activation and amplification of the cascade, whereas host cell membranes effectively inhibit further activation. For example, most mammalian cells (host cells) have high levels of sialic acid, and sialic acid favors the binding of factor H to the spontaneously deposited C3b on the cell membrane. Factor H acts as a cofactor for the serine esterase factor I, that cleaves and inactivates C3b rendering it unable participate further in complement activation. On the other hand, microbial surfaces such as bacterial cell walls, viral envelopes, or yeast cell walls have little to no sialic acid. Thus, microbial and foreign surfaces favor the binding of factor B, not factor H, to the deposited C3b. Binding of factor B to C3b allows for the interaction of factor D with factor B. Factor D cleaves a small fragment from factor B (Ba) resulting in the formation of the alternative pathway C3 convertase, C3bBb. The alternative pathway C3 convertase is stabilized by properdin, which extends the half-life of the convertase. The alternative pathway C3 convertase subsequently cleaves more C3 molecules leading to the rapid generation of C3b, and this amplification loop also amplifies the generation of C3b produced via the classical pathway and lectin pathway. The binding and cleavage of an additional C3 molecule to C3bBb forms the alternative pathway C5 convertase, C3bBbC3b, with the same function as the classical pathway C5 convertase. The alternative pathway can also be activated by the binding of properdin to molecular patterns found on pathogens or injured cells and the subsequent recruitment of fluid phase C3b.
Regardless of how complement is activated (classical, lectin, or alternative pathway), it has been estimated that the alternative pathway accounts for about 80% of generated complement activation products, and as such it is a major driver of inflammation.
The Terminal Pathway of Complement
All 3 pathways of complement activation converge at the formation of the C3 and C5 convertases, the latter of which cleaves C5. Cleavage of C5 produces the anaphylatoxin C5a, and a larger C5b fragment that initiates the nonenzymatic terminal pathway of complement ( Fig. 4 ).
The cleavage of C5 exposes a binding site on C5b for C6. The resulting C5b-6 complex then binds C7 to form C5b-7, and upon formation of this trimeric complex, amphipathic sites are exposed that allow insertion into lipid bilayers. In the event that C7 fails to bind to C5b-6, the complex is released and in the presence of C7 is able to bind to other nearby membranes. However, membrane insertion is an inefficient process, and most of the complexes are inactivated in the fluid phase. The C5b-7 complex than binds C8, exposing hydrophobic sites that form a stronger association with lipid bilayers. Finally, C5b-8 binds C9, which then unfolds in the membrane, which exposes further C9 binding sites. Thus, C5b-8 binds and polymerizes several C9 molecules, the result of which is a cytolytic transmembrane pore known as the membrane attack complex (MAC; C5b-9 n ).
Additional Pathways of Complement Activation
In addition to the classical, alternative, and lectin pathways, it is now known that the complement system can be activated via other means. The best described of such interactions is the crosstalk between the complement, coagulation and fibrinolysis pathways. As described, activated factor XII is able to activate the classical pathway of complement activation. Additionally, thrombin has been shown to both cleave C3 and also act as a C5 convertase, hence, bypassing the early pathways of complement activation. It is also well-described that plasmin and kallikrein directly cleave C3 and its activation fragments.
Relevant to the current discussion is recent data investigating complement activation in the pathophysiology of the thrombotic microangiopathies. Evidence suggests that thrombotic thrombocytopenic purpura, atypical hemolytic uremic syndrome, and typical Shiga toxin-induced hemolytic uremic syndrome are all diseases of aberrant complement activation. It is well-known that atypical hemolytic uremic syndrome results from uncontrolled complement activation secondary to mutation in complement regulatory proteins. However, until recently Shiga toxin-induced hemolytic uremic syndrome and thrombocytopenic purpura were not considered to be complement-mediated diseases. In thrombocytopenic purpura, current reports suggest widespread activation of complement as an important pathogenic mechanism. In Shiga toxin-induced hemolytic uremic syndrome, it is thought that complement is activated via p-selectin, which is upregulated as a result of endothelial damage caused by Shiga toxin.
Activation of the complement system
The Classical Pathway
The classical pathway is triggered by antibody-antigen immune complexes via C1q recognition of Fc domains in conformationally altered immunoglobulin (Ig)M or clustered IgG ( Fig. 1 ). The interaction of C1q with Fc causes a conformational change within the C1q molecule and the subsequent cleavage and activation of the associated C1r and C1s serine proteases. Activated C1s then cleaves C4 and C2 into 2 large active fragments (C4b and C2a) and 2 small soluble inactive fragments (C4a and C2b). Cleavage of C4 exposes a reactive thioester within the C4b fragment, which results in covalent attachment of C4b to the activating surface. The binding of C2 to C4b and the subsequent cleavage of C2 result in the covalently attached classical pathway C3 convertase, C4bC2a (note that there is discrepancy in the literature in the designation of C2a vs C2b). This complex cleaves C3 into C3b (large) and C3a (small). Similar to C4b, C3b contains a reactive thioester that can become bound covalently to the activating surface, and that can initiate activation of the alternative pathway. If C3b binds to the C4bC2a complex, it forms to classical pathway C5 convertase (C4bC2aC3b) that cleaves C5 into C5b and C5a, with initiation of the terminal complement pathway. Both C3a and C5a are known as anaphylatoxins and are powerful biological effector molecules with diverse functions, including inflammation, modulation of adaptive immunity, and repair and regenerative processes.
Not all antibody isotypes bind C1q and activate complement. IgM is very effective at activating complement, but some subclasses of IgG are poor activators (human IgG4 and mouse IgG1). Activation of the classical pathway can also occur independent of antibodies, and C1q can bind directly to certain microbial epitopes or epitopes exposed on apoptotic and necrotic cells. C1q can also bind to cell surfaces via pattern recognition molecules such as C-reactive protein, an acute phase protein that binds to the surface of pathogens or injured cells and activates the classical pathway. This process tags surfaces with complement proteins to facilitate their removal by phagocytes. An interaction can also occur between the coagulation cascade and the complement system, as activated factor XII is able to interact with C1 and activate the classical pathway.
The Lectin Pathway
Activation of the lectin pathway results in formation of the same C3 convertase as is generated in the classical pathway (C4bC2a). The catalytic activation sequences of the lectin pathway resemble those of the classical pathway, but with the utilization of different recognition molecules and different associated serine proteases ( Fig. 2 ). In the lectin pathway, mannose-binding lectin (MBL) or ficolins recognize patterns of carbohydrates such as N -acetylglucosamine or mannose, but not sialic acid or galactose, which provides selectivity for bacterial, viral, fungal, and parasitic cell surfaces. MBL is structurally and functionally similar to C1, and the binding of MBL (or ficolin) to a carbohydrate ligand activates MBL-associated serine proteases (MASP-1, MASP-2, and MASP-3), which generate C4bC2a in a manner analogous to that of the C1 complex.
The Alternative Pathway
In contrast with the classical pathway and lectin pathway, the alternative pathway is constitutively active and depends on the slow spontaneous hydrolysis of C3, which exposes a binding site for the alternative pathway protein, factor B, and subsequent generation of the alternative pathway C3 convertase (C3bBb; Fig. 3 ). This “tickover” process provides a primed and rapid response mechanism for the deposition of complement on foreign surfaces. Activation and amplification of the alternative pathway depend on the surface to which hydrolyzed C3 becomes attached covalently. Activating surfaces, such as the surface of a microbe, do not have the means to regulate further C3 activation and amplification of the cascade, whereas host cell membranes effectively inhibit further activation. For example, most mammalian cells (host cells) have high levels of sialic acid, and sialic acid favors the binding of factor H to the spontaneously deposited C3b on the cell membrane. Factor H acts as a cofactor for the serine esterase factor I, that cleaves and inactivates C3b rendering it unable participate further in complement activation. On the other hand, microbial surfaces such as bacterial cell walls, viral envelopes, or yeast cell walls have little to no sialic acid. Thus, microbial and foreign surfaces favor the binding of factor B, not factor H, to the deposited C3b. Binding of factor B to C3b allows for the interaction of factor D with factor B. Factor D cleaves a small fragment from factor B (Ba) resulting in the formation of the alternative pathway C3 convertase, C3bBb. The alternative pathway C3 convertase is stabilized by properdin, which extends the half-life of the convertase. The alternative pathway C3 convertase subsequently cleaves more C3 molecules leading to the rapid generation of C3b, and this amplification loop also amplifies the generation of C3b produced via the classical pathway and lectin pathway. The binding and cleavage of an additional C3 molecule to C3bBb forms the alternative pathway C5 convertase, C3bBbC3b, with the same function as the classical pathway C5 convertase. The alternative pathway can also be activated by the binding of properdin to molecular patterns found on pathogens or injured cells and the subsequent recruitment of fluid phase C3b.
Regardless of how complement is activated (classical, lectin, or alternative pathway), it has been estimated that the alternative pathway accounts for about 80% of generated complement activation products, and as such it is a major driver of inflammation.
The Terminal Pathway of Complement
All 3 pathways of complement activation converge at the formation of the C3 and C5 convertases, the latter of which cleaves C5. Cleavage of C5 produces the anaphylatoxin C5a, and a larger C5b fragment that initiates the nonenzymatic terminal pathway of complement ( Fig. 4 ).
The cleavage of C5 exposes a binding site on C5b for C6. The resulting C5b-6 complex then binds C7 to form C5b-7, and upon formation of this trimeric complex, amphipathic sites are exposed that allow insertion into lipid bilayers. In the event that C7 fails to bind to C5b-6, the complex is released and in the presence of C7 is able to bind to other nearby membranes. However, membrane insertion is an inefficient process, and most of the complexes are inactivated in the fluid phase. The C5b-7 complex than binds C8, exposing hydrophobic sites that form a stronger association with lipid bilayers. Finally, C5b-8 binds C9, which then unfolds in the membrane, which exposes further C9 binding sites. Thus, C5b-8 binds and polymerizes several C9 molecules, the result of which is a cytolytic transmembrane pore known as the membrane attack complex (MAC; C5b-9 n ).
Additional Pathways of Complement Activation
In addition to the classical, alternative, and lectin pathways, it is now known that the complement system can be activated via other means. The best described of such interactions is the crosstalk between the complement, coagulation and fibrinolysis pathways. As described, activated factor XII is able to activate the classical pathway of complement activation. Additionally, thrombin has been shown to both cleave C3 and also act as a C5 convertase, hence, bypassing the early pathways of complement activation. It is also well-described that plasmin and kallikrein directly cleave C3 and its activation fragments.
Relevant to the current discussion is recent data investigating complement activation in the pathophysiology of the thrombotic microangiopathies. Evidence suggests that thrombotic thrombocytopenic purpura, atypical hemolytic uremic syndrome, and typical Shiga toxin-induced hemolytic uremic syndrome are all diseases of aberrant complement activation. It is well-known that atypical hemolytic uremic syndrome results from uncontrolled complement activation secondary to mutation in complement regulatory proteins. However, until recently Shiga toxin-induced hemolytic uremic syndrome and thrombocytopenic purpura were not considered to be complement-mediated diseases. In thrombocytopenic purpura, current reports suggest widespread activation of complement as an important pathogenic mechanism. In Shiga toxin-induced hemolytic uremic syndrome, it is thought that complement is activated via p-selectin, which is upregulated as a result of endothelial damage caused by Shiga toxin.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


