(1)
Massey Cancer Center, Virginia Commonwealth University Health System, 1101 East Marshall Street, Richmond, VA 23298-0033, USA
(2)
Department of Human and Molecular Genetics, Virginia Commonwealth University Health System, 1101 East Marshall Street, Richmond, VA 23298-0033, USA
Cancer is a genetic disease. Cancer is distinct from most genetic conditions, however, because its associated genetic mutations are usually acquired (i.e., not inherited), and cancer involves multiple genetic mutations. A subset of cancers, however, follows classic Mendelian inheritance patterns. These hereditary cancers are the focus of this chapter.
Generally, about 5–15 % of cancers run in families. A proportion of these involve a single high risk gene usually inherited in an autosomal dominant manner. People typically have two copies of every gene — one copy inherited from mother and one from father. In an autosomal dominant condition, having inherited a single mutation on either gene copy is sufficient to lead to high risk. Being a gene mutation carrier (i.e., having inherited a mutation in one gene copy) is usually not sufficient for cancer to develop. Rather, an acquired (somatic) mutation involving the other gene copy may be necessary for cancer to develop. This phenomenon known as the “two-hit hypothesis” was classically illustrated by Dr. Alfred Knudson in the special case of hereditary retinoblastoma [1]. For most hereditary cancers, and likely all “familial” cancers, involvement of a single gene is likely not sufficient for cancer to develop. As a result, hereditary liability for cancer lies on a continuum from low risk to high-risk (Fig. 19.1).
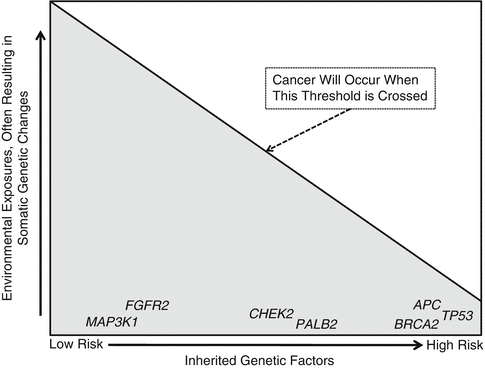
Fig. 19.1
Cancer results from a combination of inherited genetic factors and environmental exposures. High-risk genetic factors are associated with hereditary cancer, typically inherited in an autosomal dominant manner. Note that even the highest-risk inherited factors may still require additional environmental exposures before cancer occurs
The highest-risk genetic factors (at the far right of Fig. 19.1) will follow a family pattern similar to other classic inherited diseases. For hereditary cancer, the pattern is usually autosomal dominant (Fig. 19.2). This means the genetic susceptibility is shared equally among males and females, and children of individuals with hereditary cancer have a 50 % chance to inherit strong cancer risk. In contrast to sporadic cancers, these hereditary cancers occur at younger-than-typical ages (e.g., premenopausal breast cancer). Individuals with hereditary cancer risk are also more likely to develop multiple primary cancers (e.g., separate endometrial and colon cancers).
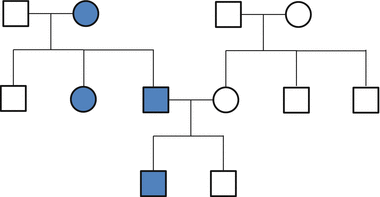
Fig. 19.2
This pedigree demonstrates a classic autosomal dominant inheritance pattern. Note that both males and females are affected. Both mothers and fathers can pass along the trait, and about half of offspring in each generation are affected
Several hereditary cancer syndromes have been described. For example, a seminal compilation of hereditary cancer syndromes from 2008 discusses 47 different conditions, many of which are associated with multiple genes [2]. In this chapter we have selected a few of these conditions that are more common and/or have characteristics that are of particular interest for molecular pathology. Specifically, we discuss hereditary cancer conditions for cancers of the breast, colon, thyroid, kidney, and pancreas, as these cancers are represented in the most common hereditary cancer syndromes. It is our hope that these examples sufficiently cover the key generalizable characteristics of hereditary cancers.
In addition to the clinical and pathology aspects of hereditary cancer, clinicians should be familiar with genetic testing technologies and test interpretation. Next-generation sequencing and the onset of multigene panel testing have brought hereditary cancer testing to new levels of complexity. Therefore, after describing several hereditary cancer conditions, we provide general information about what genetic tests to order (e.g., single-site, single-gene, multigene panel) and how to interpret test results (e.g., variant of uncertain significance).
Finally, the last section of this chapter describes the key elements of informed consent and genetic counseling. This discussion covers both the process of genetic counseling and how to access experts who can provide this service.
Hereditary Breast Cancer Syndromes
Invasive breast cancer affects about one in eight women during her lifetime and is the second leading cause of cancer death in the USA [3]. Breast cancer also affects men, although the estimated lifetime risk of 1 in 1,000 is significantly lower. The majority of breast cancer occurs sporadically with only 5–10 % attributed to inherited genetic alterations. About 25 % of the hereditary type of breast cancer is attributed to inherited, or germline, alterations in the BRCA1 and BRCA2 genes and causes Hereditary Breast and Ovarian Cancer Syndrome. Since the identification of these genes in 1994, researchers have worked to identify other high penetrance breast cancer susceptibility genes. Thus far, additional high risk genes have all been associated with known cancer syndromes like Li–Fraumeni (TP53), Peutz–Jeghers (STK11/LKB1), and Cowden syndrome (PTEN). Various other genes which are currently thought to confer a low to moderate risk of breast cancer have also been identified including CHEK2, ATM, NBS1, RAD50, BRIP1, and PALB2. Mutations in these genes likely account for the familial clustering of breast cancer that occurs in families without mutations in high risk genes (Fig. 19.3).
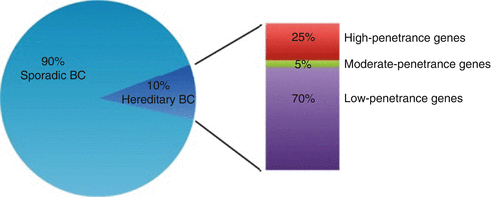
What Are the Characteristics of Hereditary Breast and Ovarian Cancer Syndrome?
BRCA1 (chromosome 17q21) and BRCA2 (chromosome 13q12.3) are tumor suppressor genes involved in double-stranded DNA repair and cell cycle regulation. Inherited mutations in these genes impair these functions and lead to a high risk for breast and ovarian cancers, as well as elevated risks for aggressive prostate cancer, melanoma, pancreatic cancer and male breast cancer. The prevalence of a BRCA2 mutation is 1 in 800 to 1 in 1,400, and the prevalence of a BRCA1 mutation is slightly higher at 1 in 450 to 1 in 800 [4]. Mutations are more common in individuals of Ashkenazi Jewish descent with about 2 % carrying one of three founder mutations: c.68_69delAG (BRCA1), c.5266dupC (BRCA1), c.5946delT (BRCA2).
Because a mutation in both copies of the BRCA1 or BRCA2 gene is required to impair DNA repair in the cell, not all individuals who inherit a mutation will develop cancer. Table 19.1 summarizes the data from various penetrance studies investigating the cancer risks to age 70 for BRCA1 and BRCA2 mutation carriers.
Occasionally, individuals inherit mutations in both copies of their BRCA1 or BRCA2 genes (biallelic). Biallelic mutations in BRCA2 cause Fanconi Anemia, an autosomal recessive cancer susceptibility syndrome characterized by progressive bone marrow failure and physical abnormalities. Biallelic BRCA1 mutations, on the other hand, are not expected to be compatible with life since one wild-type BRCA1 allele is required for embryogenesis. In 2013, Domcheck et al. validated the first example of a biallelic deleterious human BRCA1 mutation in an individual who was diagnosed with ovarian carcinoma at age 28 [8]. This individual also had short stature, microcephaly, and developmental delay. Although rare, it is possible for an individual to inherit a BRCA1 mutation from one parent and a BRCA2 mutation from the other parent. This is termed “double heterozygosity” and is more likely to occur in populations with a higher carrier frequency (e.g., Ashkenazi Jewish population). Studies suggest that these individuals have a similar probability of developing breast and ovarian cancers, but may develop these cancers at an earlier age [9].
BRCA1 and BRCA2 breast cancer tumor characteristics differ. BRCA1 tumors are usually basal-like and tend to be estrogen receptor (ESR1 also known as ER), progesterone receptor (PGR also known as PR) and erb-b2 receptor tyrosine kinase 2 (ERBB2 also known as HER2) negative (64–90 %). Given this, these tumors cannot be treated with therapies that target estrogen production or signaling or ERBB2 (HER2) targeted agents. They also have immunohistochemical features that clearly distinguish them from sporadic tumors which may help in identifying BRCA1 carriers. BRCA2 tumors, on the other hand, are typically luminal B, more differentiated and tend to be estrogen and progesterone receptor positive. However, they are not well characterized by additional immunohistochemical or morphological features that could be used to target individuals with BRCA2 mutations [10] (Table 19.2).
Table 19.2
Immunohistochemical expression of markers in BRCA1 and BRCA2 related cancers compared to sporadic cancer
Marker | BRCA1-related cancers | BRCA2-related cancers |
|---|---|---|
CK 5/6 | + | − |
CK14 | + | − |
ERβ | + | ? |
ALDH1 | + | + |
EGFR | + | + |
HIF-1α | + | ? |
p53 | + | = |
P-cadherin | + | ? |
Laminin | + | ? |
Vimentin | + | ? |
Caveolin 1 | + | = |
Bax | + | + |
BCL2 | + | + |
Active caspase 3 | + | + |
ESR1 (ER) | − | = |
CK8/CK18 | − | + |
PGR (PR) | − | = |
ERBB2 (HER2) | − | = |
FGFR1 | = | + |
FGFR2 | = | + |
Who Should Have BRCA1 and BRCA2 Testing?
In 1990, the US Department of Health and Human Services developed a set of health objectives, Healthy People 2000, aimed at improving overall health and quality of life for Americans. These objectives primarily focus on disease prevention strategies, public education and reducing health care disparities. Every 10 years, new objectives are drafted based on stakeholder feedback and the results of ongoing evaluations. Healthy People 2020 includes a genomics topic area to acknowledge the clinical utility of genetic tests and family health history in guiding health interventions (http://www.healthypeople.gov/topicsobjectives2020/overview.aspx?topicid=15). The first objective in this new area is to increase the number of women with a family history of breast and/or ovarian cancer who receive genetic counseling. This is based on a recommendation from the US Preventive Services Task Force stating that women in this high risk category could benefit from genetic counseling to learn about genetic testing for BRCA1 and BRCA2 mutations. For women who test positive, surgery could potentially reduce the risk of breast and ovarian cancer.
Genetic testing should be considered for individuals who have a personal history of premenopausal breast cancer, particularly triple negative breast cancer, and for those diagnosed at an older age with two or more close relatives with a BRCA1/2-related cancer. A comprehensive list of testing criteria has been published by the National Comprehensive Cancer Network (http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#genetics_screening). Additionally, the USPSTF provides recommendations on risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women (http://www.uspreventiveservicestaskforce.org/uspstf/uspsbrgen.htm). Many insurance companies follow these guidelines and recommendations, although variations among plans exist.
The National Comprehensive Cancer Network publishes and routinely updates evidence-based consensus guidelines regarding the management of BRCA1 and BRCA2 carriers. These guidelines include annual breast MRIs in addition to annual mammograms, chemoprevention for breast cancer, and surgical procedures to reduce the risk of both breast and ovarian cancers. (http://www.nccn.org/professionals /physician_gls/pdf/genetics_screening.pdf).
What Are Some Other Hereditary Breast Cancer Syndromes?
Less commonly, hereditary breast cancer is associated with other high risk genes related to a particular genetic syndrome (Table 19.3). In addition to increased cancer risks, these syndromes are often characterized by clinical features including macrocephaly, mucocutaneous pigmentation and colon polyps. High risk breast cancer screening (annual breast MRI and mammogram) is recommended for women with these syndromes. Increased and earlier surveillance for additional syndrome-related cancers is also recommended (http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf).
Table 19.3
Additional hereditary breast cancer syndromes
Syndrome | Gene(s) | Breast cancer risk | Additional cancers/clinical features |
|---|---|---|---|
Cowden syndrome | PTEN | 25–77 %[11] | Thyroid, uterine, and colon cancer; macrocephaly, hamartomatous polyps |
Hereditary Diffuse Gastric Cancer | CDH1 | 39–52 % | Lobular breast and diffuse gastric cancer |
Li–Fraumeni syndrome | TP53 | 50–90 % | Breast, sarcoma, brain, and adrenocortical cancer |
Peutz–Jeghers syndrome | STK11 | 45–50 % [12] | Breast, colon, stomach, sex cord-stromal tumors, uterine, lung, and pancreas cancer; mucocutaneous pigmentation |
Hereditary Colon Cancer Syndromes
About 1 in 20 individuals in the USA will develop colorectal cancer in her/his lifetime [13]. While it is the third leading cause of cancer-related deaths in the USA, that rate has been declining in recent years. This is likely due to increased colorectal cancer screening and removal of polyps before they develop into cancer. Colorectal cancers evolve over several years through the accumulation of genetic mutations that contribute to genomic instability. About 15 % of colon cancers have a familial component and another 5 % are due to inherited mutations in genes associated with well-defined genetic syndromes. Here we describe two common autosomal dominant hereditary colorectal cancer syndromes, Familial Adenomatous Polyposis and Lynch syndrome (Fig. 19.4).
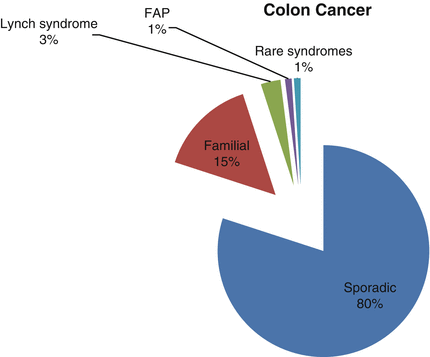
Fig. 19.4
Genetic susceptibility in hereditary colon cancer
What Are the Characteristics of Familial Adenomatous Polyposis (FAP?)
Familial Adenomatous Polyposis is caused by inherited mutations in the adenomatous polyposis coli APC gene, a tumor suppressor located on chromosome 5q21, and has a general population incidence of 1 in 8,000 [14]. Individuals with FAP develop hundreds to thousands of precancerous colon polyps between the ages of 7 and 36 years. By 35 years, 95 % of people with FAP have polyps detectable on colonoscopy. These polyps lead to colon cancer in virtually 100 % of individuals unless a colectomy is performed. Extracolonic manifestations are variably present and include: polyps of the gastric fundus and duodenum, osteomas, dental anomalies, congenital hypertrophy of the retinal pigment epithelium (CHRPE), soft tissue tumors, and desmoid tumors. Individuals with extracolonic features are sometimes referred to as having Gardner syndrome, an FAP subtype. CNS tumors, specifically medulloblastomas, characterize another FAP subtype called Turcot syndrome.
The APC gene encodes a protein that is an important regulator of epithelial homeostasis. It is also involved in controlling cell cycle progression and stabilizing microtubules, thus promoting chromosomal stability. Tumors that originate from this pathway begin as tubular adenomas and are predominantly left-sided, have a highly differentiated histology, show little lymphocytic infiltration, and are rarely mucinous.
Current molecular testing can detect APC mutations in most individuals with classic FAP (Table 19.4). Changes in this gene also cause attenuated FAP (AFAP), a form of FAP that presents with ~10–99 polyps, a later age of onset and a colorectal cancer risk to age 80 of about 70 %. Individuals with AFAP have thyroid and duodenal cancer risks similar to classic FAP; however, other extra-intestinal manifestations are rare. The AFAP colonic polyp phenotype is also similar to an autosomal recessive hereditary colon cancer syndrome called MUTYH-associated polyposis (MAP). Individuals with MAP inherit mutations in both MUTYH (mutY DNA glycosylase) gene and have colon cancer risks similar to AFAP. Extra-intestinal features including CHRPE (Congenital Hypertrophy of the Retinal Pigment Epithelium), osteomas, and dental anomalies, have also been reported. The MUTYH carrier frequency is ~1–2 %, so 1 in 2,500 to 1 in 10,000 individuals will have biallelic mutations [15].
Table 19.4
Cancer risks associated with FAP
Cancer | Risk to age 80 |
|---|---|
Colon | ~100 % if untreated |
Brain (medulloblastoma) | <1 % |
Thyroid (papillary) | 1–12 % |
Hepatoblastoma | 1–2 % by age 5 |
Pancreas | <5 % |
Gastric | <1 % |
Duodenal | 4–12 % |
For individuals with classic FAP, colectomy is recommended when more than 20–30 adenomas or multiple adenomas with advanced histology have developed; colectomy may be delayed depending on the size and number of adenomatous polyps. For individuals with attenuated FAP or MAP, colectomy may be necessary, unless the number of colonic polyps is small enough that surveillance with periodic colonoscopic polypectomy is sufficient. Screening recommendations for extracolonic manifestations are based on expert opinion. Management guidelines have been published by the National Comprehensive Cancer Network (http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#genetics_colon), the American Society of Clinical Oncology [16] and the American Society of Colon and Rectal Surgeons (http://www.fascrs.org/physicians/practice_parameters/treatment_inherited_colorectal_cancer/).
Who Should Have APC Testing?
Molecular testing for APC mutations should be considered in individuals with a personal history of more than ten adenomatous colon polyps, a personal history of a desmoid tumor, or in those with a known familial APC mutation. APC testing in individuals with the attenuated phenotype, ~10–99 colon polyps, will identify a mutation about 30 % of the time. APC testing in these cases would confirm a diagnosis and help to direct treatment and surveillance strategies. Since 20 % of affected individuals represent a do novo mutation, a dominant family history may not be evident and should not impact testing eligibility.
What are the Characteristics of Lynch Syndrome?
Approximately 15–20 % of all colorectal cancers demonstrate microsatellite instability (MSI), changes in the length of repetitive sequences throughout the genome. MSI is caused by defective DNA mismatch repair (MMR) due to either a mutation in one of the five genes involved in the DNA mismatch repair pathway, MLH1, MSH2, MSH6, PMS2, and EPCAM, or to hypermethylation of the MLH1 promoter. Tumors that exhibit MSI typically arise from conventional adenomas or, rarely, serrated adenomas (Fig. 19.5).
Fig. 19.5
Microsatellite instability in colorectal cancers
Inherited MMR defects cause Lynch syndrome, also known as hereditary non-polyposis colorectal cancer (HNPCC). Given the variety of tumor types associated with this condition, “Lynch syndrome” has become the accepted terminology. The prevalence of Lynch syndrome in all colorectal cancer cases is ~3 %; the general population incidence is estimated to be 1 in 2,000 to 1 in 660 [17]. Muir-Torre syndrome and Turcot syndrome are older terms used to describe subtypes of Lynch syndrome that include sebaceous neoplasms and/or keratoacanthomas and glioblastomas, respectively. In addition to MSI, Lynch syndrome colon tumors are predominantly right-sided, have a poorly differentiated histology, show lymphocytic infiltration, and are often mucinous.
Table 19.5 summarizes the cancer risks in individuals with Lynch syndrome which vary depending on the gene involved. Mutations in the EPCAM gene lead to silencing of the neighboring MSH2 gene and result in cancer risks comparable to MSH2 carriers [18].
Table 19.5
Lynch syndrome cancer risks up to age 70
Cancer | MLH1 and MSH2/EPCAM (%) | MSH6 | PMS2 |
|---|---|---|---|
Colon | 40–80 | 10–22 % | 15–20 % |
Endometrium | 25–60 | 16–26 % | 15 % |
Stomach | 1–13 | 3 % or less | * |
Ovary | 4–24 | 1–11 % | * |
Hepatobiliary tract | 1.4–4 | uncommon | * |
Urinary tract/renal | 1–4 | <1 % | * |
Small bowel | 3–6 | uncommon | * |
Brain/CNS | 1–3 | uncommon | * |
Sebaceous neoplasm | 1–9 | uncommon | uncommon |
Pancreas | 1–9 | uncommon | uncommon |
Biallelic germline mutations in any of the four DNA mismatch repair genes result in a childhood cancer syndrome called constitutional mismatch repair deficiency syndrome (CMMRD). This syndrome, first reported in 1999, is characterized by a variety of childhood tumors including brain tumors, lymphoid malignancies, and gastrointestinal cancer. Children with CMMRD often have skin hyperpigmentation similar to the café-au-lait spots characteristic of Neurofibromatosis type 1 [20].
In 1990, the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer organized the clinical features of Lynch syndrome families into clinical diagnostic criteria known as the Amsterdam I criteria (Table 19.6). In 1999, the Amsterdam II criteria were developed to include the other Lynch syndrome-related cancers. An additional set of guidelines, the Bethesda guidelines (Table 19.7), were established in 1996 to help guide which tumors should be tested for microsatellite instability. These were later revised in 2002 to improve sensitivity and specificity.
Table 19.6




Amsterdam criteria
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
