Fig. 12.1
The effects of chemotherapy and radiotherapy on the tumor microenvironment. A range of chemotherapeutic agents can affect the tumor microenvironment in a variety of ways. Oxaliplatin can induce immunogenic cell death in a proportion of tumor cells, which can lead to the release of tumor antigens for uptake and processing by antigen presenting cells (APC). Anthracyclines can recruit APCs and enhance their differentiation to an activated phenotype, better able to present antigen to lymphocytes. Oxaliplatin can also lead to an increased proportion of proinflammatory, M1, macrophages relative to alternatively activated, M2, macrophages. Gemcitabine, oxaliplatin and paclitaxel can reduce the frequency of myeloidderived suppressor cells (MDSC) and/or regulatory T cells (Treg) infiltrating tumors, thereby reducing their immunosuppressive effects. Tumor cells can upregulate expression of immune target molecules such as Fas and MHCI following irradiation, thereby rendering them sensitive to attack by T cells. Irradiation can also normalize dilated and chaotic blood vessels to enable T cells to access tumors. Increases in intratumoral T cells can also be achieved using antibodies against vascular endothelial growth factor (VEGF) (Adapted from Enhancing immunotherapy using chemotherapy and radiation to modify the tumor microenvironment (2013), Kershaw MH et al. [6])
12.3 Combination of Immunotherapy with Radiotherapy
Ionizing radiation induces both direct and indirect killing of cancer cells. For a long time, this treatment approach has been considered to be immunosuppressive. However, this concept has evolved over the past few years as it has been demonstrated that irradiation increases tumor immunogenicity, favoring the killing effect of an immune response against tumor cells [29]. Several studies have demonstrated that the efficacy of immunotherapy is enhanced when combined with radiation therapy [10, 30].
The term abscopal, derived from the Latin term ab (away from) and the ancient Greek term skopos (target), was introduced in 1953 to describe a rare phenomenon in which the effects of radiotherapy are observed outside the treated area [31]. Anecdotally, tumors outside the radiotherapy treatment field have been noted to shrink. This is thought to result from a putative systemic inflammatory or immune response provoked by radiotherapy. In contrast to these observations, others have described radiotherapy as an immune suppressant. Because lymphocytes are known to be very sensitive to the effects of radiotherapy, irradiation of the tumor target could potentially eliminate antitumor immune activity. This, along with the fact that there exists limited local control of radiotherapy and of early immune therapies on systemic disease, has dampened enthusiasm for pursuing this treatment combination [29].
In recent years, two case reports have highlighted the immune adjuvant effect of radiotherapy treatment in melanoma patients. In the first case, the melanoma patient had a presumed abscopal response following radiotherapy alone and a second abscopal response following a combined treatment of both radiotherapy and targeted immunotherapy [32]. In the second case, the patient initially progressed slowly despite treatment with targeted immunotherapy. This patient then exhibited a response following palliative radiotherapy with additional targeted immunotherapy treatment [33]. These initial anecdotal reports focused on the potential of radiation treatment as a mechanism to spark a systemic antitumor immune response.
Following radiotherapy, preexisting tumor-specific antibody levels were found to rise, T-cell activation markers were found to be enriched, and new antitumor antibodies were identified. Augmenting immune activity is also thought to potentiate the local effects of radiotherapy. The possibility of improving the treatment of both local and widespread disease makes this combinational treatment worthy of further investigation (Fig. 12.2).
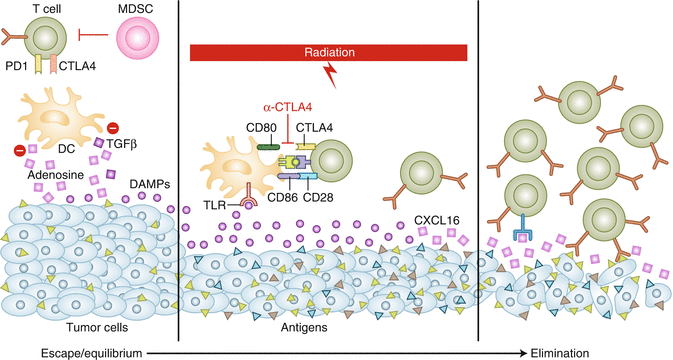
Fig. 12.2
The role of RT in induction of the antitumor immune response. At baseline, both the tumor immune microenvironment and the poor antigenicity of the tumor allow it to escape immune recognition. Targeted RT can induce increased antigenic expression, release pro-inflammatory cytokines (e.g., CXCL16) that recruit immune cells, promote antigen cross-presentation (HMGB-1 via TLR4), and induce tumor expression of death receptors. Anti-CTLA4–targeted immunotherapy can enhance the adaptive immune component by promoting antigen crosspresentation and T cell activation. Used together, RT and immunotherapy may have synergistic effects and may shift the tumor immune system balance toward elimination (Adapted from Radiation and immunotherapy: a synergistic combination (2013), Kalbasi A et al. [29])
The development of a systemic antitumor immune response has been described as concomitant immunity. Evidence suggests that radiotherapy could induce concomitant immunity where it did not exist previously (e.g., RT-induced in situ vaccination). Numerous animal models have demonstrated augmentation of systemic antitumor immunity following local radiotherapy, resulting in the reduction, control, or elimination of distant metastases, especially when used in combination with immunotherapy [32–34]. Thus, the combination of radiotherapy and immunotherapy for the treatment of locally advanced disease is currently being studied in several clinical trials, with the majority of patients being melanoma and prostate cancer patients [29, 35].
In addition to promoting recognition of tumor by preexisting tumor-specific T cells, radiotherapy has also been demonstrated to generate tumor-specific cytotoxic T cells. A recent clinical trial showed that the combination of radiotherapy with intranodal injection of low-dose rituximab resulted in the induction of systemic CD8+ T-cell immunity and regression of disseminated follicular lymphoma in immature dendritic cells and GM-CSF [12]. Irradiated tumors have been shown to upregulate death receptors (e.g., FAS), which could promote the cytotoxic effect of T cells at the tumor site. The immune system, which is devoid of its negative regulatory pathways at the site of the irradiated tumor, can function as a powerful local antitumor agent. These studies suggest a promising role for immune modulatory agents in the generation of improved radiotherapy efficacy.
Moreover, radiotherapy has been shown to cause an inflammatory response in the tumor microenvironment, resulting in the release of cytokines and chemokines and the upregulation of adhesion molecules. Recruitment of cytotoxic T cells to the irradiated tumor site is enhanced due to the release of chemokines, such as CXCL16 [36]. Irradiation was found to cause tumor vasculature remodeling, a result of the upregulation of CXCL9 and CXCL10, which enhances the density and alters the diameter of blood vessels within tumors such that they resemble capillaries. Following irradiation, T cells were found to be able to access and penetrate the tumor, inducing complete tumor regression in some cases. So, irradiation could be followed by the adoptive transfer of activated, tumor-specific lymphocytes, which previously unable to adhere to endothelium and thus access the tumor [37].
12.4 Combination of Cancer Vaccine with Immune Checkpoint Inhibitor
Immune checkpoint inhibitors have contributed great progress to cancer treatment. However, numerous challenges remain, limiting the further development of these immunotherapy drugs. Specifically, only approximately 10–30% of cancer patients with some certain types of solid tumors have exhibited an objective response to treatments using immune checkpoint inhibitors. Such challenges are attributed to properties of the tumor microenvironment (TME) [38].
The TME generates immune-tolerant conditions, posing an obvious challenge for the induction of antitumor immune responses. If the early influx of CD8+ T cells fails to clear the tumor in the TME, tumor cells which express high levels of PD-L1 will induce T-cell anergy, leading to decreased effector T-cell activity. Therapeutically blocking this pathway is thought to enable the reactivation of effector T cells in the tumor.
T cells are often the primary target of therapeutic immune checkpoint inhibitors. Effector T-cell infiltration of solid tumors is considered a signature trait of patients who have responded positively to immune checkpoint inhibitor treatment [39]. Using this marker of T-cell infiltration, it has been determined that only a fraction of solid tumor patients respond to immune checkpoint inhibitors. Thus, the remaining cancer patients would unlikely respond to immune checkpoint inhibitors when used as a single-agent treatment due to the lack of targets [40]. The TME in immune checkpoint inhibitor resistant tumors has been described to resemble that of an engine without gas. Specifically, even if the “brake” set by immune checkpoints is released with immune checkpoint inhibitor immunotherapy, no effective antitumor immune response would be elicited in this case [38].
Cancer vaccines have been demonstrated to enhance the infiltration of effector T cells into tumors in preclinical models. All vaccine-based therapies have been designed such that antigens are delivered to the patients in order to induce tumor-specific effector T cells. A vaccine-based therapy could possibly be the most efficient way to induce T-cell infiltration into tumors.
The formation of immune regulatory structures within the TME is just the first step toward the establishment of an enhanced anticancer immune response. This is attributed to the ability of lymphoid aggregates to express both effector-activating and deactivating immune signatures. Interestingly, PD-L1 expression was found to be induced in all lymphoid aggregates [41]. This observation is consistent with adaptive immune resistance, which occurs with activation of PD-L1 signaling by vaccine-induced adaptive immune response [42]. Thus, vaccine-based therapies may prime advanced tumors for anti-PD-1/PD-L1 treatments [43].
It is thought that vaccines would help stimulate the initiation of the T-cell response, while checkpoint therapies would enhance the activated T-cell response. A blockade of PD-1 or CTLA-4 combined with vaccine treatments was shown to effectively eradicate tumors in multiple preclinical models [44, 45]. Two clinical trials have been initiated which have been designed to test the pancreatic cancer vaccine-based therapy in combination with nivolumab, a treatment for advanced pancreatic cancer (ClinicalTrials.gov identifier: NCT02243371 and NCT02451982). Whether anti-PD-1 therapeutic antibodies can enhance the efficacy of cancer vaccines in the treatment of cancer remains to be studied. The combination treatment using GVAX and the anti-CTLA-4 antibody, ipilimumab, has also been demonstrated to be potentially effective in the treatment of metastatic pancreatic cancer [46]. Preclinical studies showed that the combined treatment of GVAX and ipilimumab resulted in a dramatic increase in the number of effector CD8 T cells in the tumor region, with enhanced tumor-antigen directed lytic function. These effects were found to be maximized when the CTLA-4 blockade was applied following vaccination, but not before. Moreover, the incorporation of a low-dosage cyclophosphamide treatment into this combined treatment regimen was found to provide additional preclinical benefits [47].
In summary, cancer vaccine-based immunotherapy may provide a treatment option to overcome the resistance of certain cancers to immune checkpoint inhibitors, while immune checkpoint inhibitors may further enhance the efficacy of existing cancer-vaccine therapies. Combination immunotherapy merges the strengths of each individual immunotherapy approach, with the cancer vaccine functioning to fuel the engine and the immune checkpoint inhibitors functioning to release the brake [38]. The combination of cancer vaccines and immune checkpoint inhibitors may function synergistically to induce more effective antitumor immune responses (Fig. 12.3).
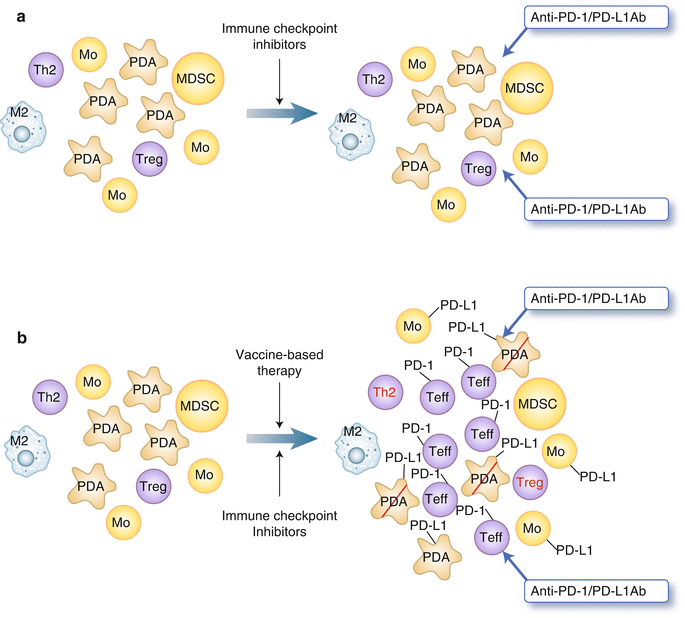
Fig. 12.3
Model for the combination of vaccine-based therapy and immune checkpoint inhibitors. (A) Pancreatic ductal adenocarcinoma (PDA) is infiltrated primarily with M2 macrophages (M2), type 2 T helper cells (Th2), myeloid-derived suppressive cells (MDSC), and regulatory T cells (Treg) but with few effector T cells (Teffs). Lacking PD-1/PD-L1 targets, PDA does not respond to single-agent checkpoint inhibitor treatments, such as anti-PD-1 or PD-L1 therapeutic antibodies (anti-PD-1/PD-L1 Ab). (B) Following vaccine-based therapy, vaccine induced Teffs are infiltrated into PDA; however, PD-L1/PD-L1-mediated immune checkpoint pathways are also induced. By targeting PD-L1/PD-L1 signals on PDA tumor cells and monocytes (Mo) induced by vaccine-based therapy, anti-PD-1/PD-L1 therapeutic antibodies enhance vaccine-induced antitumor immune responses (Adapted from Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors (2015), Kleponis J et al. [38])
12.5 Combination of ACT with Immune Checkpoint Inhibitor
Adoptive cell transfer (ACT) is thought to be a promising avenue for cancer treatment. However, despite ongoing improvements in this field, many patients do not experience clinical benefits. The tumor microenvironment is a critical limiting factor in immunotherapy that has not been fully addressed in ACT treatments. Because PD-1 has been shown to attenuate T-cell-mediated antitumor responses, blocking the PD-1 pathway in vivo could function to restore defective effector function of tumor-infiltrating T cells. Thus, an anti-PD-1 antibody could function to enhance the antitumor activity of ACT. The limited preclinical data available supports the use of combination of cellular and immune-modulating mAb therapies in syngeneic cancer models. In these models, the combination of a PD-1 blockade with murine CAR-T cells showed a significantly enhanced antitumor effect compared with either treatment method alone [48]. In addition, treatment with the anti-PD-1 antibody was found to increase expression levels of IFN-γ and IFN-γ inducible chemokine production at the tumor site. This resulted in an increased chemokine-dependent trafficking of immune cells to malignant disease sites [49]. A blockade of PD-1 in combination with ACT demonstrates therapeutic synergy and could provide a potential strategy for the improvement of clinical response rates to ACT.
We predict that, in patients with tumors containing an intermediate neoantigen-presentation capacity, the maximal immune-mediated antitumor responses would be achieved best in patients where the microenvironment can be modified and HLA-independent targeted effectors can be added, such as observed with combination immune-modulating mAbs plus ACT therapy or, potentially, CAR-T-cell therapy. However, it is important to note that a risk of toxicities could be exacerbated when these approaches are utilized. We eagerly await the results of ongoing clinical trials aimed to investigate the combination treatment of immune checkpoint inhibitors with ACT.
12.6 Combination of Various Immune Checkpoint Inhibitors
Early results from the use of combination immunotherapy strategies to block multiple immune resistance mechanisms demonstrate that a greater proportion of patients may benefit from combination therapies. Combination of anti-CTLA-4 and anti-PD-1 therapy has already shown significant clinical promise. Specifically, in a global phase III trial, combination therapy of nivolumab and ipilimumab has demonstrated an unprecedented 61% response rate and a 22% complete response rate in patients with advanced stage melanoma. These results compare with a 10–30% response rate observed in patients treated with a single agent. However, it should be noted that combination therapy was associated with a higher incidence of immune-related adverse events [50].
Based on these encouraging results, several phase I/II trials are currently underway to test the combination treatment of an anti-CTLA-4 and an anti-PD-1/PD-L1 antibody, with promising preliminary results. A phase I trial exploring the combination of tremelimumab with durvalumab treatment with GC patients is currently ongoing. An ongoing phase Ib/II trial is aimed at investigating the activity of single-agent nivolumab or nivolumab plus ipilimumab in patients with metastatic gastric cancer, pancreatic cancer, triple-negative breast cancer, and small-cell lung cancer (NCT01975831). Another ongoing phase Ib/II clinical trial of MEDI4736, a human immunoglobulin (Ig) G1κ anti-PD-L1 antibody, either as monotherapy or in combination with the CTLA-4 inhibitor, tremelimumab, monotherapy in gastric or GEJ adenocarcinoma patients (NCT02340975) is also underway.
The combination of agents that enhance effector T-cell function with agents that suppress immune-suppressive elements, such as MDSC, Tregs, and macrophages, in the tumor microenvironment may prove to be complementary, and possibly synergistic. Thus, many of these avenues are being actively investigated for cancer therapy.
12.7 Combination of Immune Checkpoint Inhibitor with Co-stimulation mAbs
The concept of increasing T-cell activity with co-stimulatory mAbs while simultaneously liberating activated T cells to lyse malignant cells by blocking PD-1 or PD-L1 is a promising antitumor approach. Numerous ongoing clinical trials involving patients across various solid tumor and hematological malignancies are being carried out to investigate this possibility.
The generation of optimal “killer” CD8 T-cell responses requires T-cell receptor activation and co-stimulation. This can be provided by the ligation of tumor necrosis factor receptor family members, including OX40 (CD134) and 4-1BB (CD137). OX40 is of particular interest as it has been shown that treatment with an activating (agonist) anti-OX40 mAb results in increased T-cell differentiation and cytolytic function. This leads to an enhanced antitumor immunity against a variety of tumor types. When given as single-agent treatments, these drugs have been shown to induce potent clinical and immunologic responses in patients with metastatic cancer. However, each of these singular agents benefits only a subset of patients. This highlights the importance of research aimed at identifying more effective combinatorial therapeutic strategies [51].
Recent data indicates that combined anti-OX40/anti-CTLA-4 mAb therapy dramatically improved survival rates observed in the poorly immunogenic TRAMP-C1 prostate and the more immunogenic MCA-205 sarcoma models. Specifically, this combination therapy was found to induce robust effector CD4 and CD8 T-cell responses required for the induction of tumor regression [52]. Marabelle et al. recently demonstrated that combined anti-OX40 and anti-CTLA-4 (with adjuvant CpG) therapy successfully induced the regression of local and distant tumors using several aggressive tumor models when the drugs were administered intra-tumorally. The mechanism by which this combination therapy functions is thought to be through the depletion of Treg cells at the tumor site, allowing for a greater inburst of CD8 T cells into the tumor [53].
Moreover, promising results from a study carried out by Guo et al. showed that the PD-1 blockade synergized with the agonistic anti-OX40 mAb to promote the regression of an implantable murine ovarian cancer, which was demonstrated to be nonresponsive to either monotherapy. This study reported that the combination therapy significantly increased the ratio of CD8 T cells present at the tumor site (peritoneal cavity), relative to both myeloid-derived suppressor cells (MDSCs) and Treg cells [54]. Another separate study showed that triple combination therapy, using co-stimulatory anti-OX40 and anti-4-1BB mAbs along with an inhibitory anti-PD-1 mAb, was also effective in a murine hepatocellular carcinoma model, with enhanced tumor infiltration of cytotoxic effector T cells observed [55].
In the near future, more data will be available that will enable the evaluation of the toxicity-benefit ratio of different immunotherapeutic combinations. We anticipate that this knowledge will provide new insights, leading to the design of novel therapies for advanced stage gastric cancer.
12.8 Combination of Molecularly Targeted Agents with Immunotherapy
Molecularly targeted agents provide selectivity and are the cornerstone for “precision medicine.” However, while targeted agents are associated with high effective rates, patients are known to often develop resistance to these drugs inevitably. Immunotherapies exploit the endogenous immune system of patients to eradicate tumors, providing a mechanism for sustained treatment resulting in long-term treatment-free survival in some patients. Combination treatment regimens provide the sustained clinical benefit of immunotherapy along with the rapid and high response rates provided by molecularly targeted therapy. This combinatorial approach could potentially offer both short- and long-term benefit to cancer patients. In this vain, numerous efforts have been initiated to combine molecularly targeted agents (MTAs) with immunotherapy.
The use of targeted agents that efficiently kill tumor cells, initiating the concomitant release of tumor antigens, should activate a specific immune response. It is possible that strategies bringing together immune checkpoint agents and MTAs would result in the induction of an immune memory, with all of its benefits. If this is the case, it would lead to a more sustained inhibition of tumor growth than would be achieved with either treatment modality alone. A great interest has developed in gaining an understanding of how immune checkpoint inhibitors can be used in conjunction with molecularly targeted agents to improve clinical outcomes for patients with advanced stage cancer. The advent of novel immunotherapeutic and molecularly targeted agents has provided numerous effective options for the treatment of cancer. However, at the same time, this has added a level of complexity in determining the most appropriate initial treatment plan for each individual patient [56].
In a phase I trial comprised of patients with BRAF V600 mutation-positive melanoma aimed at evaluating concurrent treatment with vemurafenib and ipilimumab, dose-limiting hepatotoxicity was observed, with further patient accrual to the study stopped [57]. Future studies aimed to optimize doses, timing, and other factors could result in lower, more manageable toxicity of these drugs. Several different approaches could be considered for evaluation. Results from an additional phase I study provided evidence of both clinical activity and a manageable safety profile for an anti-PD-L1 antibody used either in combination with dabrafenib and trametinib in BRAF mutation-positive melanoma patients, in combination with trametinib in BRAF wild-type melanoma patients, or following trametinib in BRAF wild-type melanoma patients [58]. While these data are encouraging, longer follow-up studies to include the duration of response data are necessary to properly assess these treatments. Information gained from clinical studies in melanoma patients may be translatable to gastric cancer patients.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


