Fig. 11.1
Erosion on the surface and lymphoid aggregates that mainly composed of mixed inflammatory cells, cryptitis, and crypt abscesses within the lamina propria are seen in inflammatory polyp (HE ×100)
Treatment: Choi et al. [2] reported that polyps showed regression in two patients with giant pseudopolyps associated with ulcerative colitis and Crohn’s disease. Giant pseudopolyps require surgical treatment in the presence of obstruction, intussusceptions, or hemorrhage [2].
Inflammatory Myoglandular Polyp
Inflammatory myoglandular polyp was first described by Nakamura et al. [12] in 1992. It is a nonneoplastic colorectal polyp with three main histological features: (1) hyperplastic glands with cystic dilatation, (2) inflammatory granulation tissue in the lamina propria, (3) proliferation of smooth muscle from the muscularis mucosa. It most commonly occurs in the sigmoid colon, rectum and other areas of the colon, and very rarely reported in cecum and anal channel [12–15]. The most common symptoms are occult or overt bleeding, hematochezia, and associated development of anemia [13, 14, 16]. Men are affected more often, and it has a wide range of age (15–78 years) [12].
Its pathogenesis is controversial, and chronic trauma and mucosal prolapse have been proposed as etiologic possibilities [12, 14]. Cases with concomitant diverticular disease have also been reported [12, 16]. Some authors consider these polyps as a type of hamartoma.
Macroscopically, they range from 0.4 to 2.5 cm in size [12]. Most are pedunculated; however, sessile cases have also been reported with rounded, smooth, or lobulated surface [12, 13]. Having mucin-filled cross sections, hyperplastic crypts are typical, and also smooth muscles are radially arranged like branches of a tree towards the surface. Smooth muscle can be immunohistochemically stained with actin in order to demonstrate smooth muscle fibers [13]. On the surface, luminal fibrinoinflammatory exudate, serrated or hyperplastic changes of the epithelium and regeneration are observed. In mucosal erosion and hemorrhagic areas, hemosiderin deposition and granulation tissue are seen within the lamina propria. Juvenile polyps, inflammatory fibroid polyps, cap polyps, Peutz–Jeghers polyps should be considered in differential diagnosis. Juvenile polyp should be considered first in differential diagnosis. Juvenile polyps develop in younger ages, and the surface includes ulcerated, dilated cystic glands in the lamina propria, inflammatory infiltrate of neutrophils and eosinophils [17, 18]. The findings including presence of varied-sized blood vessels in the stroma, a fibrous connective tissue consisting of onion-skin-like spindle-shaped fibroblasts wrapping these vessels, and presence of nonspecific inflammatory cells are in favor of inflammatory fibroid polyp [19]. Inflammatory fibroid polyp most often occurs in the stomach and small intestine, rarely in large intestine [19]. The surface of cap polyps presents with a characteristic “fibrin cap” appearance of intense inflammation, dilated, elongated, or tortuous hyperplastic colonic crypts and granulation tissue [20, 21]. Peutz–Jeghers polyps are mainly located in the small intestine, and rarely in large intestine, and they are characterized with branching of smooth muscle fibers from the trunk and projecting towards the normal glands on the surface [22, 23].
Prolapsed-Type Inflammatory Polyps
The term mucosal prolapsed syndrome was first used by Boulay et al. [24] combining solitary rectal ulcer syndrome and related conditions. However, prolapsed-type inflammatory polyp often develops as a result of localized protrusion of mucosa unrelated to the solitary rectal ulcer syndrome.
Its pathogenesis remains unclear, but it leads to twisting of mucosa due to damaged peristalsism as a consequence of traction, distortion, and trauma, which results in torsion of blood vessels, and localized ischemia and tissue damage, and ultimately lamina propria fibrosis during repair [25, 26]. Depending on the anatomic location of the injury and underlying causes, these polyps can be also called inflammatory cap polyps, colitis cystica polyposa, diverticular disease-associated polyps, and inflammatory cloacogenic polyps of the anal transitional zone. They usually show histological abnormalities overlapping with that of mucosal prolapse [25]. Furthermore, solitary or multiple polyps, which share common histopathological characteristics with mucosal prolapse, can also be called prolapse-induced inflammatory polyps or prolapsed-type polyploidy mucosal folds [25, 26].
The classic histological features of prolapse-induced inflammatory polyp are as follows: (1) a variable degree of fibromuscular hyperplasia of the lamina propria, chronic inflammation, ulcer, reactive epithelial change, telangiectatic vascular malformations; (2) thickening, widening, and vertical extension of the muscularis mucosa into the lamina propria; and (3) crypt abnormalities (e.g., elongation, hyperplasia, architectural distortion, and serration) [24–26].
Inflammatory Cap Polyp (Cap Polyposis)
It was first defined in 1985 by Williams et al. [20]. Colonic dysmotility and mucosal prolapse are reported as the etiological factors [21]. It is more common in females, with a wide range of age (12–76 years) [21]. Clinical signs may include primarily mucous diarrhea, followed by bloody stools, abdominal pain, constipation, and rarely rectal bleeding, epigastric pain, and nausea [20, 21]. The most common location is rectum or rectosigmoid colon, but rarely it also involves the descending colon [27, 28].
Macroscopically, it is a small, sessile, or semi-pedunculated polyp, ranging from 0.3 to 3 cm in diameter [29].
Histologically, it is a nonneoplastic polyp characterized by large, elongated, or tortuous hyperplastic colonic crypts, and a characteristic “cap” appearance of intense inflammation, ulceration, and granulation tissue in the lamina propria. Some polyps may have smooth muscle fibers and fibrosis in relation with the prolapse etiology [21, 29].
Treatment: Cap polyposis may regress after treatment for ulcerative colitis or nonspecific colitis [21]. Surgical resection is required for many patients with multiple polyps, while it may spontaneously regress in some patients [29, 30] or may regress with treatment for H. Pylori [31, 32]. Recently, there are some reports indicating that infliximab is an effective therapy as well as [33, 34] some other publications concluding that infliximab is ineffective for treatment of cap polyposis [35].
Colitis Cystica Profunda
Colitis cystica profunda is a rare benign condition characterized by mucin-filled mature cystic dilated crypts in submucosa, sometimes in muscularis propria [36, 37]. Colitis cystica profunda was first described by Virchow [37] in 1863 for multiple polypoid lesions with submucosal cysts. In 1957, the term colitis cystica profunda had started to be used in association with submucosal cysts and polypoid lesions. Colitis cystica profunda often occurs in patients with solitary rectal ulcer syndrome, inflammatory bowel disease, diverticular disease, and postirradiation strictures and infectious diseases [36, 38–42]. Patients may present with clinical symptoms including rectal bleeding, abdominal pain, nausea, and vomiting [36, 38–41]. The most common site is rectum [42, 43].
The pathogenesis is considered to be related to vascular injury and ischemia associated with prolapse, torsion, and trauma of the mucosa and submucosa [36].
Macroscopically, it may present as a focal, diffuse, or segmental lesion [36]. Focal or segmental lesions can mimic invasive adenocarcinoma or accompany adenocarcinoma [40].
Histologically, multiple dilated cysts are seen in submucosa, sometimes in muscularis mucosa or subserosa, with mucin-filled crypts surrounded by non-desmoplastic stroma. Consequent displaced glands show such a lobular nature that it is distinguished from the infiltrative features of irregularly shaped glands in adenocarcinoma. The epithelium of these displaced crypts shows normal or reactive changes. Pseudostratification and slightly increased mitotic activity are observed. Loss of nuclear polarity, increased nuclear to cytoplasmic ratio and atypical mitosis should suggest adenocarcinoma. Rare cases together with adenocarcinoma and tubular adenoma are reported [40, 44]. Furthermore, cases concomitant with cap polyposis are also reported [41].
In many patients with rectal prolapse, colitis cystica profunda may regress with treatment of defecation disorders (avoidance of constipation, high-fiber diet, laxatives) [45]. Patients may undergo surgical resection who have obstruction, severe symptoms, concomitant IBD and who are confused with carcinoma [32, 36, 40, 41].
Diverticular Disease-Associated Polyp
In 1985, Franzin et al. [46] reported that mucosal polypoid lesions can be seen in diverticular disease. Similarly, Mathus-Vliegen and Tytgat [47] described polypoid lesions in mucosal prolapse syndrome in eight patients with diverticular disease. It was also described by Kelly [48] as “polypoid prolapsing mucosal folds in diverticular disease” in patients with diverticular disease. It is a rare nonneoplastic polyp characterized with a muco-submucosal elongated polyp in colon, particularly in sigmoid colon [26, 46, 48]. Clinical symptoms such as bloody stool and abdominal pain may occur [26, 49, 50].
Macroscopically, it appears as a bright red, polypoid or slightly bulging mucosa with a smooth and soft surface in the sigmoid colon with diverticulus [46–48]. It measures around 0.5–3 cm in size [49, 50].
Histologically, early lesions show vascular congestion, hemorrhage, and hemosiderin depositions. Advanced lesions may present with edema of lesions, capillary thrombus, lamia propria fibrosis, and changes in crypts such as dilatation and branching. More advanced lesions may show leaflike changes, serrated hyperplastic changes of the epithelium, smooth muscle prolapse into the lamina propria (α smooth muscle actin can be used to demonstrate it by immunohistochemical analysis [26]), crypt hyperplasia, and mucin depletion [48]. These polyps look like inflammatory cap polyps, but the surface of cap polyps are covered with cap-like fibroids and an extensive granulation tissue, with less telangiectatic vascular changes compared to prolapse-type polyps [26]. Epithelial hyperplasia should not be mistaken for adenoma, and rare pseudosarcomatous changes (large, hyperchromatic myofibroblasts in the stroma) should not be mistaken for sarcoma [48].
Inflammatory Cloacogenic Polyp
Inflammatory cloacogenic polyp (ICP) was first described in 1981 [51], and it is often associated with mucosal prolapse, and sometimes with hemorrhoids. It is a rare benign inflammatory polyp occurring around the anal transitional zone and rectum [52]. ICP can be accompanied by adenocarcinoma, villous adenoma, tubular adenoma, and Crohn’s disease [53]. Patients with ICP may present with complaints such as rectal bleeding, tenesmus, anal mass, and a palpable mass [52, 54]. The ICP mostly occurs in women, during their fifth to seventh decades of life [55].
Macroscopically, it ranges from 1 to 5 cm in diameter in the form of an irregular or round mass [52, 54].
Microscopically, surface of the polyp is typically eroded, and covered with a squamous epithelium or transitional zone. Without any dysplastic features, elongated and tortuous crypts protrude into the submucosa in a complex tubulovillous growth pattern. Thin, longitudinal and circular strands of the proliferated fibromuscular stroma along with smooth muscle fibers of the muscularis mucosa extend into the lamina propria among the crypts. In stroma, edema, vascular ectasy, inflammatory cells and granulation tissue are observed [54]. Human papillomavirus (HPV)-positive and dysplastic cases of the squamous epithelium have also been reported [56]. In differential diagnosis, solitary rectal ulcer syndrome should be ruled out first; solitary rectal ulcer occurs in younger age and is localized in the rectum [57]. Since it may display similar clinical and endoscopic characteristics with malignant colorectal polyps, it should be differentiated histologically [58].
Hamartomatous Polyps
The term hamartoma refers to overgrowth of normal tissues in irregular fashion, forming a nonneoplastic mass in their natural anatomic locations [59]. Hamartomatous polyposis syndromes (HPS) are rare, genetic, and characterized by development of hamartomatous polyps in gastrointestinal tract (GIT) [60, 61]. HPS includes juvenile polyposis syndrome (JPS), PTEN-hamartoma tumor syndromes (Cowden syndrome, Bannayan–Riley–Ruvalcaba and Proteus syndrome), and Peutz–Jeghers syndrome [60].
Juvenile Polyps and Juvenile Polyposis Syndrome (JPS)
Juvenile polyps are seen in four different settings: (1) sporadic or isolated juvenile polyps of the colon, (2) infantile JPS, (3) juvenile polyposis coli (JPC), and (4) generalized JPS [62].
Sporadic or isolated juvenile polyps often occur in rectosigmoid colon in 2 % of children or adolescents, with no potential for malignancy [63–65]. On the other hand, risk for malignancy is high in juvenile polyposis [59, 62]. JPC and generalized JPS are differentiated by the location and extent of polyps [59, 66]. In JPC, polyps occur only in the colon, while generalized JPC involve stomach, small intestines, and colon [59, 66]. They represent subtypes of the same condition, which is a familial rare disease with autosomal dominant transmission, occurring in the first two decades of life [59]. Infantile JPS presents with clinical symptoms including rectal bleeding, diarrhea, and exudative enteropathy often during the first 2 years of life, with a high mortality [66, 62]. There are also various accompanying organ malformations (umbilical fistula, malrotation of small intestines, unilateral renal agenesis, Fallot tetralogy, and aortic stenosis) [59]. If it is symptomatic in adults, usually there is also acute or chronic rectal bleeding, prolapse, and diarrhea [67].
The diagnostic criteria for juvenile polyposis are as follows [59, 61, 62]:
1.
Evidence of three or more juvenile polyps (the number may vary between 3 and 10 polyps) by colonoscopy
2.
Juvenile polyps involving the entire GIT
3.
Any number of juvenile polyps and a familial history of JPS
Germline mutations of SMAD4 tumor suppressor gene located in chromosome 18q21 and bone morphogenetic protein receptor type 1A (BMPR1A) involved in BMP signal pathway located in the chromosome 10q22-23 were reported to be responsible for development of JPS [59, 62, 66–69]. PTEN mutation is controversial. Recent studies suggest that PTEN mutations are associated with Cowden syndrome rather than JPS [62, 66, 67].
Histopathologically, juvenile polyps are characterized with hyperplasia of mucous glands, retention cysts, edema of the lamina propria, loss of smooth muscle, and inflammatory infiltrates of many lymphocytes and plasma cells and rare neutrophils and eosinophils in stroma [59]. In differential diagnosis, Cronkhite–Canada syndrome (CCS) should be considered, which is histologically very similar. Microscopically, the polyps in both syndromes cannot be differentiated; however, in CCS pathological changes such as edema in mucosa of the colon, cystic dilated glands, and increased inflammatory cells are noted; whereas the intervening mucosa between juvenile polyps is normal in JPS [70]. Furthermore, clinically ecdodermal abnormalities, such as alopecia, nail dystrophy, and skin hyperpigmentation are noted in CCS [71].
Polypectomy is an adequate treatment in solitary juvenile polyps due to absence of any potential malignancy [64, 65]. However, JPS has a high malignancy potential, with no extraintestinal malignancy, contrary to other hamartomatous syndromes [59, 62]. Carcinoma was reported in colon, stomach, and rarely in pancreas and duodenum/ampulla [68]. Therefore, JPS requires endoscopic screening and prophylactic surgery, if appropriate, in those with a high risk of CRC [67]. If any of SMAD4 and BMPR1A genes are identified, family screening should be done. Those with a negative genetic result should receive the same screening program with normal population, while colonoscopy and upper GIT endoscopic screening are recommended every 3 years for those who have a positive result or present with symptoms before 15 years of age [62, 67]. Juvenile polyps should be endoscopically removed, and endoscopic screening should be repeated every year until complete removal of polyps [67]. If the result is negative, then a repeat screening should be performed every 3 years [67]. If the polyps are too much to be removed by endoscopy, then a surgical procedure (partial colectomy, subtotal colectomy, total proctocolectomy) is indicated [67]. Similarly, subtotal or total gastrectomy can be considered against risk of cancer when it occurs in the stomach [67].
PTEN-Hamartoma Tumor Syndromes
Cowden Syndrome (CS)
Cowden syndrome (CS) is a very rare autosomal dominant familial hamartoma/neoplasia syndrome [59]. In CS, the most common involvement sites are ectoderm and endoderm layers although all three germ cell layers are affected by hamartomas, and almost all patients have pathognomonic dermomucosal lesions (trichilemmomas, acral keratoses, oral papillomas, mucosal lesions) [59, 60, 67]. Breast lesions (fibroadenomas, fibrocystic disease, and adenocarcinoma) are more common in women, and cases of breast carcinoma were reported in men as well [72]. The risk for breast carcinoma in women is 30–50 %, of which 25 % are bilateral [59]. Thyroid abnormalities may include multinodular goiter, adenoma, and rarely carcinoma [67, 73]. The risk of carcinoma is increased by 10 % compared to normal population [59]. Increased risk for macrocephaly, cerebellar gangliocytoma, genitourinary malformations and endometrial carcinoma, as well as renal cell carcinoma and melanoma has been reported [60, 67].
In CS, germline mutations in tumor suppressor gene PTEN (phosphatase and tensin homologue, deleted on chromosome 10) located in chromosome 10q22-23 were reported [59].
The cystic, dilated mucin-filled crypts within lamina propriety in hamartomatous polyps of CS cannot be histologically differentiated from that of juvenile polyps [61]. Hamartomatous polyps may occur in stomach, small intestines, and/or colon. Glycogenic acanthosis of the esophagus is very common [59]. In addition, colonic lipoma, fibrolipoma, fibroma, ganglioneuroma, and adenoma have been reported [67].
Although the risk of CRC in CS remains unknown, other organs with a risk of malignancy should be evaluated. It is recommended that women should undergo mammography and transvaginal ultrasound and endometrial biopsy beginning at age 30, and both genders should be examined by thyroid ultrasound, renal ultrasound, and colonoscopy after 40 years of age [60].
Bannayan–Riley–Ruvalcaba Syndrome (BRRS)
In 1990 Cohen suggested that Ruvalcaba–Myhre–Smith syndrome, Bannayan–Myhre–Smith syndrome, and Riley–Smith syndrome are phenotypically and genetically similar, and unified them as BRRS [74]. Marsh et al. identified an association between PTEN gene and BRRS [75]. It is indicated that CS and BRRS have mutation of the same PTEN gene, even within the same exons; however, the phenotypic difference between these two syndromes can be explained by the hypothesis that there is a distinct expression in the PTEN gene [76].
Clinically, macrocephaly, hemangiomas of varying size, lipomatosis, genital pigmentations, hamartomatous intestinal polyps, and lipid myopathy may occur in BRRS [60, 67, 73]. The polyps in GIT and those of JP are morphologically similar, and if they occur, extraintestinal evidence is required for diagnosis of CS/BRRS [61]. One or more extraintestinal manifestation with or without polyps or a familial history of CS/BRRS can be considered as the main diagnostic criterion, although it is uncertain [61]. Patients with BRRS should undergo the same surveillance programs as patients with CS [60].
PTEN-Related Proteus Syndrome and Proteus-Like Syndrome (PS)
It is classified in the group of PTEN hamartoma syndrome [60]. There is a disproportionate growth of body systems, which may affect skeletal, skin, and central nervous systems [60, 77]. In addition to PTEN mutations, somatic activation in AKT1 gene in PS was also reported [78]. Periodic ophthalmologic evaluations and brain MRI are recommended for follow-up of these patients [77].
Peutz–Jeghers Syndrome (PJS)
PJS is characterized by mucocutaneous melanosis, hamartomatous polyposis of the GIT, and increased risk of intestinal and extraintestinal malignancy [60, 61]. It is an autosomal dominant inherited syndrome, with an incidence between 1:8,300 and 1:200,000 [79]. The patients may present with rectal bleeding, intestinal invagination, anemia, and mucocutaneous pigmentation during early childhood or infancy [60]. Mucocutaneous pigmentation is present in approximately 95 % of patients, occurring on the extremities, nose, and around the eyes, but mainly on the lips, buccal mucosa (Fig. 11.2), and perianal area [60, 61]. Polyps are mostly in the small intestine and colon and may vary from one to hundred in numbers [60].
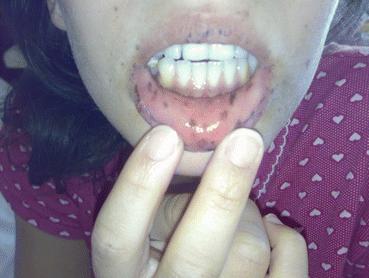
Fig. 11.2
Pigmentation on lips and buccal mucosa
PJS diagnosis is based on clinical presentation, histopathological characteristics of polyps, family history, and genetic testing. Diagnostic criteria include: (1) two or more histopathological PJ polyps (PJP), (2) familial history in an individual with any number of PJP, (3) mucocutaneous pigmentation in an individual with a familial history, (4) mucocutaneous pigmentation in an individual with any number of PJP [60, 80].
Development of PJS is associated with germline mutations in the serin-threonine kinase 1 (known as STK11, LKB1) located in the chromosome 19p13.32 [60, 61, 79, 80].
Histologically, polyps originate from elongated glands, and branching of smooth muscles of muscularis mucosa [60]. Lamina propria may be normal or cellular. Distinct from JPs, the ratio of mucosa to lamina propria is normal, and there are no cystically dilated glands in the epithelium (Fig. 11.3). These polyps may also be found in extraintestinal sites such as urinary bladder, gallbladder, ureter, and bronchi [81].
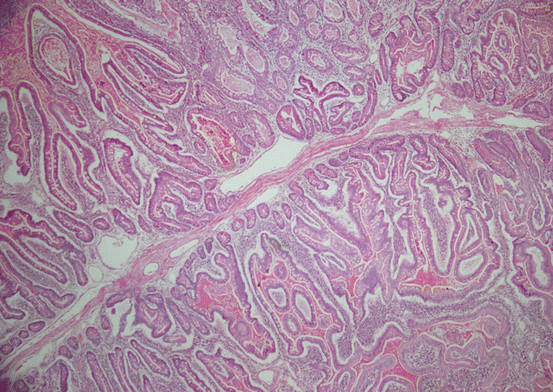
Fig. 11.3
Peutz–Jeghers polyp is composed of complex arborizing fronds and typically non-dysplastic, elongated glands (HE ×100)
In PJS, an increased risk for intestinal and extraintestinal malignancies is reported [60, 61, 80] There is also an increased risk for GIT cancers, mainly colon, and pancreas, stomach, small intestine, and for extraintestinal malignancies such as lung, and cervix, ovary, uterus, mainly breast in women, and testis in men [61, 80, 82].
The recommendations in the PJS surveillance program include colonoscopy, endoscopy, and double-contrast radiological study every 2–3 years beginning at the age of 18, and in the presence of a polyp, removal of the polyp; mammography, gynecological examination, pap smears in women, and testicular examinations and ultrasound in men [60]. However, there are some views advocating that these screening programs should start earlier, for example at the age of 8, and even much earlier in symptomatic patients [80]. As well as other views emphasizing that screening program should start at different ages based on each organ, and they should be more frequent every 1–2 years [61].
Epithelial Polyps
Colorectal Serrated Polyp Family
Hyperplastic polyps are known by their benign cytology and serrated architecture, and traditionally have been considered as nonneoplastic lesions with no malignant potential [83]. However, larger polyps located in the right colon were termed mixed hyperplastic/adenomatous polyps in 1984 by Urban et al. [84]. In 1990, Longacre et al. [85] identified neoplastic nature of these lesions, and coined the term serrated adenoma. The terminology of serrated polyps is still controversial [86, 87]. According to WHO, a recent morphological classification of these polyps with a serrated architecture can be divided into two groups based on presence or absence of dysplasia [88]. The subtypes of serrated polyps include hyperplastic polyp (HP), sessile serrated adenoma/polyp (SSA/P), sessile serrated adenoma/polyp (SSA/P) with dysplasia (DSSA/P), and traditional serrated adenoma (TSA) (Table 11.1). Others suggest that the term adenoma should be used interchangeably with dysplasia [89]. Based on this, it is indicated that the term SSP should be used for lesions with pseudo or nonordinary dysplasia so that the term adenoma can be used synonymously with dysplasia in the gastrointestinal tract. It is suggested that serrated polyps with true dysplasia should be considered as “traditional serrated adenoma,” and since the term sessile serrated adenoma is considered to be premalignant by clinicians, alert them to follow-up the patient as in adenoma [89].
Table 11.1
Classification of serrated polyps according to WHO
Type of Polyp | Presence of dysplasia |
|---|---|
Hyperplastic polyp (metaplastic polyp) | |
MVHP | No |
GCHP | No |
MPHP | Type A, but in reactive form |
SSA/P | No |
SSA/P with dysplasia (mixed hyperplastic adenomatous polyp-MHAP) | Yes |
TSA | Yes |
Serrated polyposis | Dysplasia as the disease progress |
Molecular Features of the Serrated Carcinogenesis Pathway
Majority of colorectal cancers (CRCs) are known to develop from ordinary adenomas through chromosomal instability molecular pathway [90]. Nonetheless, approximately 15 % of CRCs are characterized with high-frequency microsatellite instability (MSI-H) caused by defective deoxyribonucleic acid (DNA) mismatch repair (MMR) [90, 91]. MSI was first described in relation to Lynch syndrome (hereditary nonpolyposis colorectal cancer) that results from mutations in MMR genes [90]. Hypermethylation of cytosine-phosphorothioate-guanine (CpG) islands of MLH1, promoter of methylator phenotype (CIMP) results in hypermethylation of MLH1 MMR gene, leading to defective MMR, and eventually to MSI [90]. Methylation of the promoter regions in CpG islands of DNA repair genes (MLH1 and methyl guanine methyl transferase- MGMT) and suppressor genes (p16, p14, THSBS1) is also considered to be the main mechanism in progression to sporadic MSI-H carcinomas [90, 92, 93]. Thus, CIMP seen in almost all cases with sporadic MSI and those who are microsatellite stable (MSS) in CRC [90]. CIMP has a strong association with point mutations in BRAF [94]. Mutations in BRAF gene result in activation of the protein kinase pathway that promotes cell proliferation and survival [90]. About 40 % of CRCs with MSI and MLH1 hypermethylation carry oncogene mutations in BRAF [90].
Recently, with the identification of serrated neoplasia pathway, it has been agreed that some types of hyperplastic polyps may also evolve to malignancy [91, 96]. Thus, it was proposed that through this pathway hyperplastic or hyperplastic-like polyps (serrated polyps) could progress to adenocarcinoma of dysplastic sessile serrated adenoma/polyp (DSSA/P) and MSI-H [94–99]. Histological classification of serrated polyps is based on comparison of molecular features of the main polyp subtypes [90]. It is believed that many CRCs with CIMP phenotype develop via the serrated neoplasia pathway [100]. This pathway activates the mitogen-activated protein kinase (MAPK) pathway as a result of BRAF mutation in the colorectal mucosa, leading to increased expression of p16INK4a and secretion of insulin-like growth factor binding protein 7 (IGFBP7). p16INK4a or IGFBP7 methylation leads to development of SSA/P [99, 101]. A definite relationship was found between the serrated polyps, particularly SSA/P and MSI and the sporadic CRCs which promote CIMP [102]. CIMP was defined as a specific marker for development of MVHP and advanced serrated polyp/adenoma [90, 103]. CIMP often occurs in SSA/P located in proximal colon and polyps with an SSA/P histology at the margins of MSI CRCs [86, 104]. hMLH1 methylation occurs at a later stage in serrated polyps, leading to dysplasia [94]. Furthermore, advanced age, proximal location, poor differentiation, mucinous histology, high BRAF mutation, and low p53 mutation rates have also been found to contribute to the development of CIMP-H CRCs [93].
Genetically, CIMP-H CRCs can be divided into two groups: those with MSI and BRAF mutations and the second with MSS and KRAS mutations [93]. BRAF mutation and negatively correlated KRAS mutations are also reported to be associated with development of colorectal adenocarcinomas [91]. BRAF V600E mutation occurs at an early stage of pathway, and often found in MVHP and SSA/P with high level CIMP [86, 94]. BRAF and KRAS mutations are early molecular alterations in serrated lesions. No BRAF mutation is observed in ordinary adenomas [105]. Neither BRAF mutations nor CIMP have been found in CRCs with germline MMR mutations that result in Lynch syndrome [94] These findings highlight that SSA/Ps are precursors of sporadic MSI CRCs.
A second alternative pathway of carcinoma development is associated with KRAS mutations in CIMP-low and microsatellite-stable cancers [93–95, 99]. The precursor lesion of this pathway is GPHP/TSA [90, 99, 101]. Recent reports have indicated that KRAS mutation in normal mucosa activates Wnt signal pathway, leading to TSA [101]. KRAS mutations are common in rectal and polypoid TSAs, but rare in SSA/Ps [106]. In this presumed serrated pathway, inhibition of repair gene MGMT DNA by promoter hypermethylation in development of carcinogenesis has been thought to associate with KRAS mutation and CIMP-low status [94]. Thus, despite increased methylation in TSA, it is absolutely not methylation of MLH1, and it is not associated with CIMP-H MSI carcinomas [88]. Such carcinomas are reported to have poorer prognosis [95].
Two serrated neoplasia pathways segregate to KRAS or BRAF mutations based on lesion type, either polypoid or flat, respectively [90, 96]. There are data available suggesting that about 30 % of colon adenocarcinomas is derived from the serrated neoplasia pathway [101]. These pathways are summarized in Fig. 11.4 [90, 95, 96, 101].
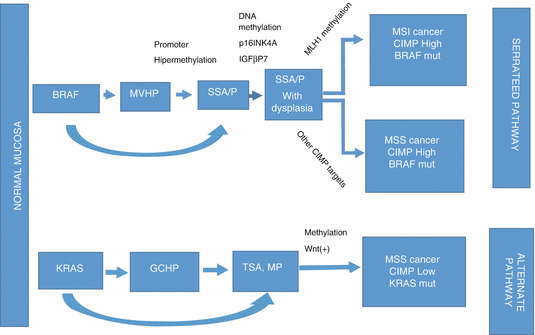
Fig. 11.4
Potential two serrated neoplasia pathways are illustrated. The upper diagram shows cancer formation with high methylation while the lower diagram shows alternative cancer formation with low-methylation. The upper serrated neoplasia pathway shows development of SSA/P or de novo SSA/P from MVHP, eventually leading to MSI/MSS cancers with CIMP-high (CIMP-H) and BRAF mutations. In the lower alternative serrated pathway precursor lesions are GCHP and TSA. It shows formation of CIMP-low (CIMP-L) and KRAS mutation cancers
Nowadays, various factors involved in the tumorigenesis of CRC have been investigated. For example, estrogen receptor alpha (Erα), being expressed in normal colorectal mucosa, and secreted frizzled related protein (SFRP)1 genes as well as CpG island methylation are suggested to be associated with tumorigenesis [107]. ERα and SFRP1 expressions were reported likely to be associated with adenoma, and hyperplastic polyp/serrated adenoma; however, study failed to show any association [107].
Hyperplastic Polyps (HPs)
Clinical Features
HPs account for 10–15 % of all colon polyps, and the most common (80–90 %) of all serrated polyps [1, 96]. While “traditional hyperplastic polyps” were regarded as a distinct entity, now they are considered as a subgroup of serrated polyps [1, 90]. These polyps can be found throughout the entire colon, but they are more common particularly in distal colon and rectum [83]. The incidence increases with age [108]. It occurs in asymptomatic individuals older than 50 years, and more common in women [109]. Although lifestyle, alcohol consumption, nutritional factors, low folate intake, and obesity are mostly associated with adenoma, they are also reported to be associated with hyperplastic polyps [109]. A study by Wallance et al. reported that CpG island methylation was associated with folate levels, and showed that individuals with low folate intake had an increased methylation in normal colon mucosa, leading to carcinoma [107]. As these lesions are usually asymptomatic, and they are frequently detected incidentally during colonoscopy. In some cases, these lesions cannot be differentiated endoscopically from adenomas, and thus they require biopsy and cauterization [1, 83].
Pathological Features
Macroscopically, the majority of hyperplastic polyps measure less than 0.5 cm, and are mostly found in rectum and distal colon [110]. The hyperplastic polyps greater than 2 cm have a small risk of dysplasia and malignant transformation [83].
Microscopically, all types are characterized with a nondysplastic appearance, displaying an irregular sawtooth or luminal serrated contour on upper half of the crypts mostly, with a tubular architecture at the base [97, 98]. These polyps show a simple tubular architecture (decrescendo pattern) with a combination of goblet and mucous cells, or only goblet cells without any branching and budding or a crypt dilatation [110]. Tubules have a perpendicular and symmetrical pattern, without any horizontal elongation or branching at the base [98]. In HPs, there is a regularly thickened basal membrane beneath the surface epithelium that is not common in SSAs [111]. Proliferation area is confined to the lower part of the basally located crypt, and wider than normal mucosa at the crypt as detected by Ki67, immunohistochemically [112, 113]. In hyperplastic polyps there is a strong expression of MUC2, and possible expression of MUC5AC and MUC6 [114]. Recent studies have shown AMACR- and p16-positive at the basal portion of the crypts in hyperplastic polyps of the colon, and AMACR was also found to be positive in the colorectal carcinomas of this region [115, 116].
A study by Torlakovic et al. [83] classified left-sided hyperplastic polyps into three groups based on their morphological growth pattern, and lack of proliferation or maturation: microvesicular cell-type, goblet cell-type, and mucin-poor-type. Microvesicular hyperplastic polyp (MVHP) is the most common one, and considered to represent a “typical” hyperplastic polyp by many pathologists [83]. It frequently involves the left colon [87, 117]. Compared to a normal mucosa, it is characterized by abundancy of vesicular mucin/vesicular cells and scanty number of goblet cells (Fig. 11.5). These lesions are more common in the left colon, and characterized by expanded proliferative regions covering more than basal half of crypts and pronounced luminal serration [83]. Nuclear atypia is variable, mild nuclear atypia is seen in most of the polyps, but severe atypia is rare [83]. Dystrophic goblet cells and cells with round vesicular nuclei or with prominent nucleolus are usually rare, and if present, they are located in the lower part of the crypt. Most importantly, these lesions display surface maturation, while traditional serrated adenomas have no characteristic eosinophilic cytoplasm certainly. There may be focally concentrated hypermucinous cells in the superficial part. However, the cells have a mature appearance with small nuclei, but no hyperchromasia or atypia. The deep regenerative part of the microvesicular hyperplastic polyps appear hyperchromatic compared to the normal cells of colonic crypts, which may be confused with adenoma. However, hyperchromatic regenerative part has a regular maturation towards surface. Muscularis mucosa shows thickening and extension into the lamina propria [83].
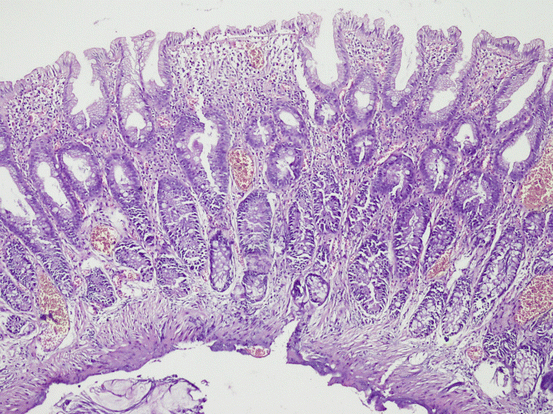
Fig. 11.5
Hyperplastic polyp, microvesicular type showing luminal serration and mixture of columnar and goblet cells (HE ×100)
Rarely, large microvesicular hyperplastic polyp may have a slight structural distortion or slight cryptic dilatation. In this case, some researchers believe that microvesicular hyperplastic polyp is a precursor of the sessile serrated polyps. Although rare, an increase in mitosis and in Kulchitsky cells (eosinophilic neuroendocrine cells) may occur [83]. Neuroendocrine cells with clear cytoplasm can be observed between the proliferative and nonproliferative zones in the mid-one-third of the crypts [83]. Immunohistochemistry has shown CK20-positivity in the epithelial surface, which occupies a larger area than normal mucosa, and Ki67-positivity at the base of the crypts [106]. BRAF mutation and DNA methylation abnormalities were common in cases with microvesicular hyperplastic polyp [86, 90, 91, 101, 110, 117]. BRAF mutation was identified in 76 % of cases as a specific marker, and DNA methylation (CIMP) was less common than serrated adenomas in 47 % of cases. MGMT loss was found in a small percent (26 %), KRAS in 13 %, and MLH1 in 39 % [91]. A study reported that specific antibody VE1 which allows detection of BRAF V600E mutation immunohistochemically was expressed specifically in the serrated areas of microvesicular hyperplastic polyps, and staining was stronger in serrated polyps [117].
Goblet cell hyperplastic polyps (GCHP) represent the second most common type of hyperplastic polyps, and they are usually small sessile lesions, smaller than 0.5 cm. They are more common in left colon [87, 98]. These polyps show elongated crypts with no microvesicular mucin but rich in goblet cells. Normally they are sessile, however, mostly confined to the surface and upper portions of the crypts, with very few luminal serrations [83]. Nuclear atypia and mitosis are usually not seen [83]. There is no or minimal nuclear stratification [83]. They show typical morphological findings of HP including extension of mucosa, thickened basal membrane and muscularis mucosa, and increased Kulchitsky cells [83]. Clear cell neuroendocrine cells are found very rarely [83]. In GCHP, typically DNA methylation (CIMP) is rare, and TP53 mutations are often negative, and there is no or little APC mutation [105]. In these polyps, KRAS mutation is dominant (42 %), while BRAF mutation is 21 % [87, 110].
Mucin-poor hyperplastic polyps are the least common type [83, 110]. Some authors believe that they are derived from microvesicular hyperplastic polyps resulting from inflammation, mucin depletion, and irritation due to injury and prolapse [98]. These lesions have a nonpedunculated, regenerative and nonstratifying appearance, consisting of small cells that have narrow cytoplasm and more hyperchromatic nuclei than MVHP [98]. They show a micropapillary architecture, increased clear cell neuroendocrine cells, mitosis, decreased Kulchitsky and goblet cells, but increased irregular, dystrophic goblet cells with prominent nuclear atypia [83]. Inflammation in the lamina propria is typical. KRAS mutation can be found in mucin-poor hyperplastic polyps [117] In a study by Kim et al. [93] KRAS, BRAF mutations were observed in 25 % and CIMP in 75 % respectively.
Differential Diagnosis
Hyperplastic polyps should be differentiated from TSA, SSA/P and prolapse-type inflammatory polyps.
Conventional adenomas show hyperchromatic, pseudostratified, cigar-like atypical nuclei. Adenomas show misplaced glands as well as atypical features such as many mitoses, even single-cell necrosis [88].
TSAs are easily differentiated from HPs, with pronounced eosinophilic cytoplasm of the cells throughout the crypt, their larger and villous structure, serration at the basal of the crypts, and presence of dilatation and branching [98, 110, 118, 119].
HPs might need to be differentiated from SSA/Ps due to presence of focal serrated areas, particularly in MVHPs [98, 119]. Unlike HPs, SSA/Ps show elongated crypts, marked serration throughout the crypts, reversed “T” or “L” shaped elongated, horizontal and branching crypts at the bottom, increased dystrophic goblet cells, focal nuclear stratification, and mitosis on the upper portion of the crypts [98, 118, 119].
Natural History and Treatment
Until recent times, definitive treatment has not been considered for small hyperplastic polyps, and they were removed by biopsy since they are not differentiated from adenomas during endoscopy [1, 83]. However, there is epidemiological and morphological evidence showing that HPs, particularly microvesicular type, rarely progress to carcinoma due to their natural course and biologic characteristics [86, 120]. In fact, there is increasing evidence that the risk of progression to cancer should be determined depending on the number, size, and location of “hyperplastic” polyps rather than the specific morphologic subtypes [121]. It was reported that the risk for colorectal carcinoma increased in HPs that were larger than 1 cm, multiple, and located in the proximal colon in the serrated pathway of carcinogenesis [97, 121–123]. In this respect, it is suggested that a large hyperplastic polyp located in the proximal colon should be completely removed, and colon should be examined thoroughly for any potential synchronous advanced neoplasia, and such patients require follow-up [97, 118].
Sessile Serrated Adenoma/Polyp (SSA/P)
Clinical Features
Unlike left-sided “hyperplastic” polyps, SSA/Ps are often located on the right side of the colon (71–75 % at the proximal splenic flexure), and larger than hyperplastic polyps, and nonpedunculated [91, 110, 124, 125]. Also, these right-sided polyps were reported to occur more often in smokers and with a family history of CRC compared to those with left colon localization [125]. Endoscopically, these lesions often show a regular surface covered with yellowish mucus [97, 126]. Some polyps resemble the surrounding intact mucosa, and show regularly arranged circular dots that can be easily distinguished from adenomas by high-resolution chromoendoscopy; however, they look like MVHP [88, 126]. As mentioned before, molecularly SSA/P, CpG island methylation (CIMP) MSI, and MSS are considered as precursors of sporadic carcinomas [90, 101, 110]. In SSA/Ps, the main characteristics of BRAF mutation include loss of MLH1 gene expression due to promotor hypermethylation and nuclear translocation of β catenin [110]. It shows a high rate of DNA methylation (CIMP) (75 %) and BRAF mutation (80 %), but a low rate of KRAS mutation (6 %) [91].
Pathological Features
Morphologically, these polyps show elongated crypts, epithelial serration, and crescendo type of dilatation, which is seen more clearly in the base of the crypt [110]. The base of the crypt shows an irregular inverted T-shape or an L-shape form, reduced amount of lamina propria between crypts, and an inverted growth pattern beneath the epithelial surface [87, 110]. Crypts may rarely show herniation through muscularis mucosa leading to “pseudoinvasion” or “inverted growth pattern.” Although its pathogenesis remains unknown, these features may also be present in HPs, but they are more common in SSA/Ps [98]. Due to goblet cell or gastric foveolar cell differentiation at the base of the crypt, proliferative zone is asymmetrical, and often not located at the base of crypt [111]. Presence of two or three consecutive crypts with these features can be interpreted as SSA/P [110]. Immunohistochemically SSA/Ps are characterized by CK20-positivity in epithelial surface and irregular and scattered distribution of Ki67-positive cells at the base of the crypt, while basal part of some crypts may have CK-positivity instead of Ki67-positivity [106]. Furthermore, there may be increased expression of gastric-type mucins, including MUC5AC and MUC6 [88, 110, 114]. SSPs may show nuclear atypia in many stages and in every part, but especially in the upper one-third of the crypt [88]. There is little or no stratification at the one end of the spectrum, low rate of mitosis, and marked superficial maturation [87]. SSA/P may show focal cytologic dysplasia. As with conventional adenoma, low or high degree dysplasia or progression to carcinoma may be observed [97]. Furthermore, cuboidal cells with open vesicular-type chromatin and marked nucleus and increased cytoplasmic eosinophilia at the other end of the spectrum may be seen but is rare (Fig. 11.6). These polyps are more aggressive than ordinary adenomas [97]. These findings suggest a diagnosis of TSA, but, there are no long filiform or villous projections contrarily [96, 110]. SSA/P and TSA are usually easy to distinguish, but they can be described as “serrated polyp, unclassified” when differentiation is not possible [88].
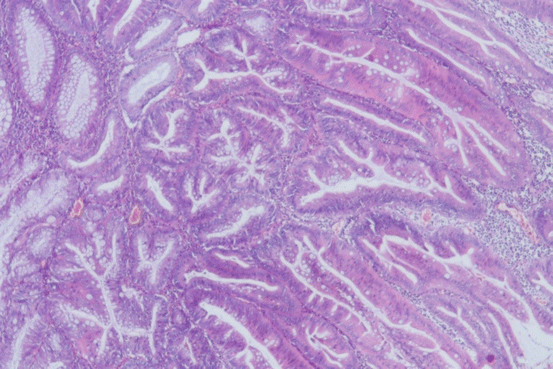
Fig. 11.6
SSA/P shows an area of marked cuboidal cells and eosinophilic cytoplasm of high-grade dysplasia (HE ×200)
Natural History and Treatment
Unfortunately, the natural history and malignancy potential of SSA/Ps still remain unclear, and studies on these subjects are ongoing [87, 90, 127, 128] Right-sided polyps greater than 1 cm were reported to have a high risk of becoming cancerous [123]. Another study reported that the risk for development of cancer in right-sided polyps was 20–30 % [129]. While there are some studies suggesting that the risk for progression to carcinoma was mostly related to the size of the lesion, some studies have indicated that dysplasia, loss of DNA repair, and recurrent MSI even in small SSA/Ps might result in a faster progression to neoplasm [87, 127, 128]. It is reported that it takes about 15 years for SSA/Ps to become cancerous [128].
All serrated lesions between the sigmoid and proximal colon and those measuring greater than 0.5 cm with rectosigmoid location should be completely removed during colonoscopy [98]. The unremoved polyps should be reevaluated at an interval of 2–6 months, and completely removed upon examination of the clinician and the pathologist [130].
Dysplastic Serrated Polyps
Dysplastic Sessile Serrated Polyp or Mixed Hyperplastic Adenomatous Polyp (MHAP)
Clinical Features
In the literature, the polyps in this group are given different names such as “mixed hyperplastic polyp- ordinary adenoma (HP-TA)” and “mixed SSA/P-TA” [87]. Although they have been referred to as such in the past, now it has been recognized that these polyps occur as a result of dysplastic changes within a sessile serrated polyp, not simultaneous growth of hyperplastic and adenomatous elements [87]. Unlike ordinary adenoma, they do not show any APC gene mutation [88]. Several studies have found that KRAS, BRAF, MSHI-H, and CIMP mutations are 22–43 %, 40–75 %, 0–5 %, and 50 %, respectively [120, 131, 132].
Pathological Features
Morphologically, these lesions show microvesicular type of changes of the hyperplastic polyp, but more often they have features of SSA/P or ordinary adenoma along with dysplastic changes [98]. In fact, some polyps have both conventional and serrated adenomatous features, and ectopic crypts may be focal as in TSA [106]. Dysplastic areas appear like tubular adenoma or tubulovillous adenoma, with a low- or high-grade dysplasia [98]. They often architecturally resemble the traditional adenoma, which often shows cigar-shaped stratification with hyperchromatic nucleus, mitosis, and amphophilic cytoplasm [98]. Another form of dysplasia, serrated dysplasia, may occur [88]. The cells are cuboidal in shape, have eosinophilic cytoplasm, vesicular chromatin, large, round nuclei, and prominent nucleolus, with mitotic activity [98]. Serrated dysplasia has a higher risk of progression to carcinoma [88].
Natural History and Treatment
Sessile serrated polyps with dysplasia are more aggressive than ordinary adenomas, and it is important to remove them completely due to hypermethylation and microsatellite instability, and risk of progression to carcinoma even though it is rare [87]. Total excision can be achieved endoscopically or surgically. Endoscopy should be repeated in these patients within 1 year to make sure that the lesion was completely removed and there is no progression. Afterwards, it can be followed up as a conventional adenoma.
Traditional Serrated Adenoma (TSA)
Clinical Features
TSA is much less common than SSA/P or HP, and accounts for less than 1 % of all colorectal polyps [110, 133]. Occurring in the left colon, it is often located in rectosigmoid, well confined, and protruding from the surface [87, 89, 106]. Endoscopically, it may be sessile, flat, and polypoid [89]. It looks pedunculated rather than sessile [134]. The mean age of onset is 60–65 years, which is typically older than patients with hyperplastic polyp or sessile serrated polyps [85]. It is more common in females [134].
Since there is confusion in the literature in referring to many different types of lesions as serrated adenoma, the clinical, epidemiologic, and molecular features of these rare lesions remain unknown. Several studies found that KRAS, BRAF, and CIMP mutations were 0–28 %, 60–76 %, and 43–80 %, respectively [105, 120, 131], while only one study found that MSI-H was 3 % [131]. However, recently it has been widely accepted that carcinoma mainly develops from KRAS mutation [95, 101]. Recent studies showed that KRAS mutation is dominant in TSA in the serrated neoplasia pathway and may develop de novo or from GCHPs [101]. TSAs may show methylation, but no MLH1 hypermethylation, and they are not associated with CIMP-H MSI carcinomas, but effective in development of MSS and CIMP-L carcinomas [88, 101].
Pathological Features
Macroscopically, it looks like an ordinary adenoma [110]. Microscopically, TSA is characterized by confluent cells having a serrated papillary or elongated finger-like villous growth pattern with pink eosinophilic cytoplasm [96, 110]. In TSA, epithelial surface may show distinct forms [106]. In some areas, the tip of these villous projections may be conjoint, protruding to the surface in the form of a tennis racquet (micropapillation), and even though focal, it may have a smooth surface as in SSA/P [87, 106] (Fig. 11.7). Goblet cells may be seen, pronounced in some cases [88, 98]. TSAs may show two types of dysplasia; ordinary adenoma-like and serrated-like [88]. Dysplasia may show nuclear atypia, nuclear stratification, cells with hyperchromasia, and intense hypereosinophilia in all layers of the epithelium, more prominent in the epithelial surface [87, 98, 119]. These cells have a reduced nuclear to cytoplasmic ratio, coarse chromatin, vesicular nucleus, and obscure nucleolus [106]. Unlike ordinary adenomas, mitosis is rare [106, 110]. Premature crypt formation perpendicular to the longitudinal axis of the villi, called ectopic crypt formation, is a typical feature of TSAs [135] (Fig. 11.8). They show proliferative activity with prominent nucleolus, vesicular, and hyperchromatic nucleus [97]. Therefore, immunohistochemical analysis shows that ectopic crypts have strong CK20 staining at luminal surfaces, and Ki67-positivity in deeper portions [87, 106].
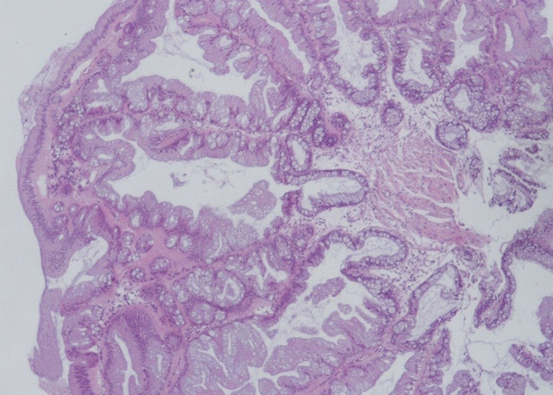
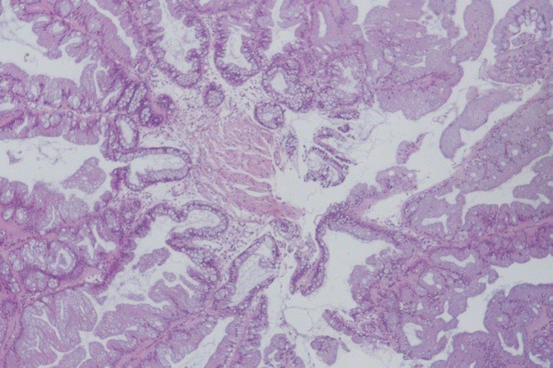
Fig. 11.7
TSA shows confluent tips of villous projections in the epithelial surface with pronounced serration in all layers (HE ×100)
Fig. 11.8
Pronounced serration, goblet cells in between and ectopic crypt formations (HE ×100)
Natural History and Treatment
Natural history and malignancy potential of TSAs still remain unclear. A study of filliform serrated adenomas by Yantiss et al. [134] found high-grade dysplasia in 22 % and invasive adenocarcinoma in 6 % of polyps. They also found that 33 % of polyps showed hyperplastic or sessile serrated polyp morphology in adjacent regions, suggesting that these are possible cancer precursor areas [134]. The rate of malignant transformation is similar to that of traditional adenomas [136]. In this respect, it is reported that TSAs, with or without dysplasia, should be treated like ordinary adenomas, and total excision is essential with surveillance intervals similar to that of traditional adenomas [87]. The risk of progression to CRC is reported to be related to the size and location of the polyp [97]. The rate of progression to CRC is faster in larger polyps located in proximal colon compared to those in distal colon [97].
Serrated Polyposis Syndrome (SPS)
Clinical Features
The term “hyperplastic polyposis” was first coined in 1980 by Williams et al. [137]. Serrated polyposis was first described in 1996 by Jeevaratnam et al. [138] as familial serrated polyposis syndrome. Although it is commonly recognized as hyperplastic polyposis, morphology of the polyps is often in the form of SSA/P [139]. However, since hyperplastic polyps can be found in combination with TSA, sessile serrated polyps with dysplasia and traditional adenomas, the term “serrated polyposis” is now widely preferred [88, 140]. SPS is commonly diagnosed at a mean age of 52–56 years [99, 140–142]. Although some publications report that it is more common in females, others indicate that it occurs most commonly in males [140, 142]. So there is no gender superiority [99]. It is often asymptomatic; however, complaints of bleeding can be seen in large polyps [97]. Endoscopically, the polyps are often smaller than 0.5 cm, sessile, and rarely large and pedunculated [140]. Small polyps are frequently scattered throughout the colon, while the large polyps are located in proximal colon [88]. There are two clinical types: Type 1 includes numerous large SSA/Ps, dysplastic sessile serrated polyps, HPs and even traditional adenomas, with significantly increased risk of cancer [101]. Type 2 includes numerous (usually smaller than 0.5 cm) HPs (with microvesicular and/or goblet cell), with slightly increased risk of cancer [88].
Genetically, its etiology and molecular features remain unknown. Although there are indications that those with many polyps in younger age have familial history, and it is a genetic disease [143], most of the patients are sporadic [98]. Some studies indicated that serrated neoplasia pathway and BRAF mutation may be effective in development of carcinoma in patients with SPS [99, 120, 142, 144]. Since MUTYH-associated polyposis shows multiple SSA/P similar to SPS, it is proposed that MUTYH gene might have also been involved in development of SPS [140, 145]. It is suggested that Type 1 may be associated with BRAF mutation, and Type 2 with KRAS mutation [88].
Pathological Features
According to the recent WHO criteria, any of the following criteria should be present for a diagnosis of SPS: (1) at least five serrated polyps located in proximal of sigmoid colon, two of them must be larger than 10 mm; (2) any number of serrated polyps located in proximal of sigmoid colon in person with first-degree relatives with diagnosed SPS; or (3) more than 20 serrated polyps distributed throughout the colon [88].
A recent study compared immunohistochemical staining patterns of p53 among polyps of SPS and SSA/P, HP, and ordinary adenomas, and found that polyps were stained for SSA/P [139]. Polyps rarely show MVHP morphology, and are greater than normal in size. Cytologically, carcinoma may develop more rapidly in dysplastic SSA/P [88, 97, 101].
Natural History and Treatment
Although the risk for CRC is certainly unknown in SPS, many studies indicates the increased risk [98, 99, 140, 142, 143]. A retrospective study indicated that the risk for CRC was increased by 7 % in patients on surveillance [142]. Other reports have indicated that the incidence of CRC is increased during diagnosis in patients with SPS, and CRC may develop in 25–70 % of patients during surveillance [99, 140, 142, 143]. The CRC in these patients are located predominantly in the proximal colon [140, 143].
Treatment for SPS is variable, and not standardized [142]. Early and frequent endoscopic follow-up and surgical colectomy are among the treatment options. Both CRC and SPS risks are increased in patients with polyp who have a first-degree relative with SPS, and therefore they should undergo colonoscopy every 5 year or more frequently [98, 99, 142]. If colonoscopic surveillance is not appropriate, then right hemicolectomy or total colectomy is recommended [98]. Colectomy is recommended for multiple polyps, particularly in the proximal colon [98]. In conclusion, the treatment should be individualized based on the age of the patient, comorbidity, number and size of polyps, type of polyp, and presence of dysplasia and carcinoma [88]. The risk for adenocarcinoma is remarkably increased in untreated patients with SPS [97, 101].
Conventional Adenomas
Clinical Features
Although adenomas are almost always asymptomatic, overt or occult rectal hemorrhage may occur in many patients [146, 147]. The incidence of adenoma often increases with age. It is most common in the fifth decade of life [146, 148, 149]. The risk for having at least one adenoma after the age of 50 years was reported to be around 30–50 % [147]. While some studies indicate that it is more common in males [150, 151], other studies do not support it, and report that it is more common in females [152, 153]. High levels of Vitamin D and calcium were reported to reduce the risk for colorectal adenoma, particularly those in the proximal colon [154, 155].
Molecularly, adenomatous polyposis coli (APC) gene involved in development of adenomas is a tumor-suppressor gene located on the chromosome 5q21-22 [156, 157]. Numerous pathogenic mutations (somatic and genetic) in the pathway including β-catenin and APC protein complex were reported to be involved [158]. β-catenin is involved in cell adhesion binding via E cadherin, playing a pivotal role in the Wnt/Wg signal pathway [158]. In normal cells, the level of cytoplasmic β catenin is downregulated by APC-mediated degradation [159]. If the APC protein is not functional, β-catenin level rises in the cytoplasm, enters the nucleus, enabling transcription of many oncogenes including c-myc through Wnt/Wg signal pathway [160]. Thus, inactivation of normally functioning APC/β-catenin complex results in epithelial proliferation in dysplastic epithelium from the base of the crypts, and development of adenoma [156]. Additional mutations and loss of heterozygosity inactivate the tumor suppressor gene p53 and SMAD2 and SMAD4 on chromosome 18q, leading to carcinoma [101, 161]. As a result, large TA and TVA are originated from TA and they proceed to CRC with CIMP negative and KRAS mutation, respectively [101] (Fig. 11.9).
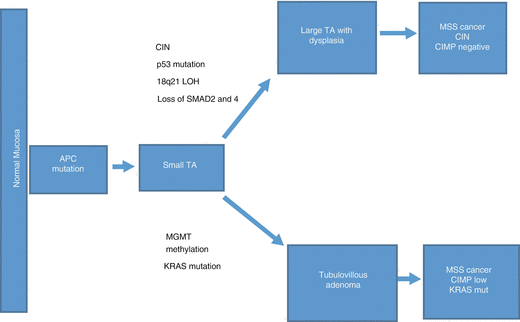
Fig. 11.9
A schematic diagram of molecular changes in development of adenoma – CRC (CIMP CpG island methylator phenotype, CIN Chromosomal instability, MSS Microsatellite stable, TA Tubular adenoma)
The adenomatous polyps of the colon are categorized as TA, TVA, and villous adenoma (VA) based on their architectural nature. Basically, they are classified as pedunculated or sessile/flat (nonpedunculated), and pedunculated polyps are more common [162]. However, some authors reported that most of the polyps were endoscopically sessile (87 %) [153, 163]. Most of the polyps have TA morphology, followed by TVA, and VA, respectively [153, 163, 164]. Some studies reported that TVA was more common [151]. Polyps are often located in the left colon [151, 163], mostly measuring 0.5–1 cm in diameter [151, 153] (Fig. 11.10).
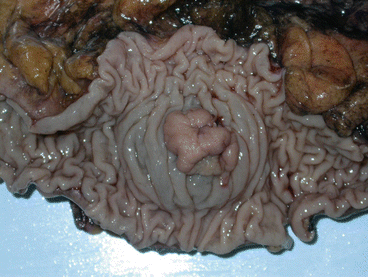
Fig. 11.10
A macroscopic view of TA with a diameter of 2 cm located in the sigmoid colon
Pathological Features
Adenomas are morphologically defined as dysplastic clonal proliferation of colonic epithelium. Being microscopically categorized as tubular, tubulovillous, or villous, adenomas are differentiated based on a reasonable rule; villous lesions contain at least 75 % villi, while tubular lesions have less than 25 % [165, 166] (Fig. 11.11). In some reports, the rates have been given as 80 % and 20 %, respectively [156, 167]. Tubulovillous lesions contain between 25 and 75 % villi [165, 166] (Fig. 11.12). The rate of villous differentiation increases with the increasing size of adenoma. Villous lesions represent an important parameter in diagnosis of advanced adenomas [89, 130, 164, 168–171].
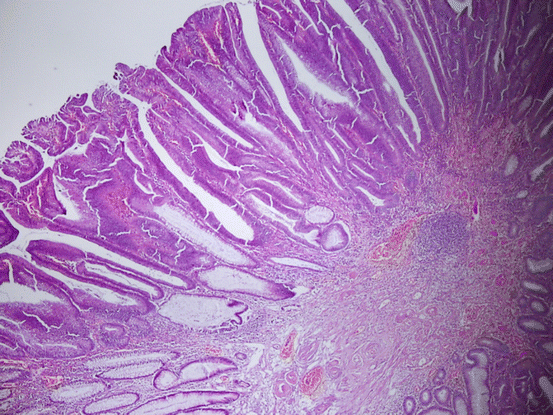
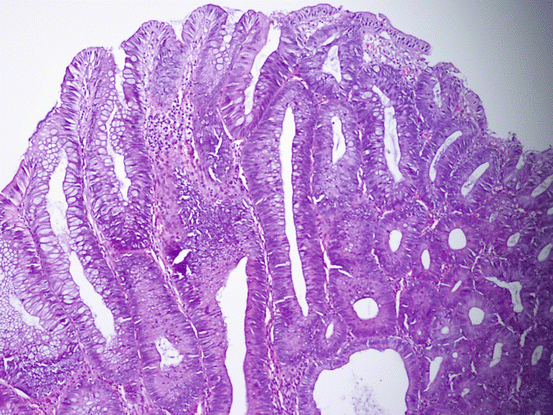
Fig. 11.11
Villous adenoma with low-grade dysplasia (HE ×40)
Fig. 11.12
Tubulovillous adenoma with low-grade dysplasia (HE ×100)
Crypt apoptosis, neutrophilic infiltrate of the lamina propria and crypts (focal active cyrptitis) are common in conventional adenomas [153]. Neutrophilic infiltrate of the lamina propria can be seen without focal active cryptitis [153]. This finding is evident particularly in high-grade adenoma and mainly in invasive areas [153]. Some adenomas, particularly pedunculated adenomas, may show dilated and ruptured crypts with mucin extravasation into the lamina propria. This feature is often associated with misplacement of the epithelium into the submucosa (will be discussed further) [89, 172, 173].
Dysplasia was previously classified in three groups as mild, moderate, or high [167]. At present, a dual classification is used as low- (mild or moderate) and high-grade (severe, including in-situ carcinoma) intraepithelial dysplasia [165, 174]. The advantages of this classification are as follows: (1) it reduces interobserver variability in interpretation of dysplasia by pathologists; (2) it provides clinical pathological compliance for surveillance and treatment options; and (3) it prevents misinterpretation of the term “in-situ carcinoma” as indicative of a malignant behavior, and unnecessary resections of the colon.
Microscopic definition of adenoma includes at least a low-grade of dysplasia. Some data report that high-grade dysplasia is more common, and they mainly have TVA and VA morphology [151]. Although it was reported that high-grade dysplasia and carcinoma were more common in flat adenomas, with a higher malignant degeneration, it is still controversial [165, 175, 176]. The size of the polyp is not associated with dysplasia; sometimes even small adenomas may show high-grade dysplasia [168].
Low-grade dysplasia is defined as the nuclei that are pseudostratified or partially stratified and reach lower half of the cell cytoplasm in architecturally noncomplex crypts and epithelium [156]. Mitotic activity can be seen, but atypical mitosis, significant loss of polarity and pleomorphism are minimal. Crypts are arranged in parallel to the surface. High-grade dysplasia is defined as marked pseudostratification or stratification of the neoplastic nuclei, which extend into the luminal aspect of the cytoplasm, show significant pleomorphism, increased mitotic activity, significant increase in nuclear to cytoplasmic ratio, and loss of polarity [167] (Fig. 11.13). As architectural changes, back-to-back arrangement of glands can be seen [167]. As it progress to neoplasia, glands lose their previous structure, becoming more irregular and complex. In addition, neoplastic nuclei become paler and nucleolus gets prominent.
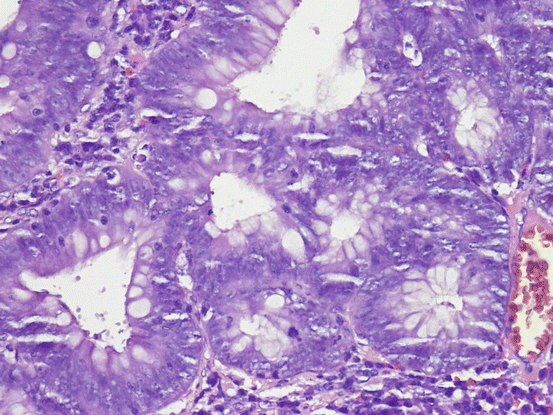
Fig. 11.13
Crypts show significant pseudostratification and stratification, pleomorphism in cells, increased mitotic activity, increased nuclear to cytoplasmic ratio and loss of polarity (HE ×400)
Advanced adenoma refers to adenomas that are larger than 1 cm with a villous structure and high-grade dysplasia [89, 130, 164, 168–171]. Advanced adenomas have a high risk of CRC, and it is emphasized that these patients require close clinical surveillance [89, 130, 169–171].
Single-cell infiltration, small gland proliferation, or significant enlargement of back-to-back glands with cribriform architecture, and their invasion into lamina propria or muscularis mucosa accompanied with desmoplasia is defined as intramucosal adenocarcinoma [165] (Figs. 11.14 and 11.15). For the differentiation of intramucosal adenocarcinoma from high-grade dysplasia, intramucosal carcinomas show significant invasion into lamina propria and it is accompanied by desmoplasia [165]. Invasive carcinoma occurs with penetration beyond muscularis mucosa and invasion to the submucosal peduncle [165] (Fig. 11.16). Several studies have shown that high-grade dysplasia, even adenomas including intramucosal adenocarcinoma, has no metastatic potential [162]. Thus, if these lesions can be removed with intact margins, polypectomy alone is an adequate treatment [162].
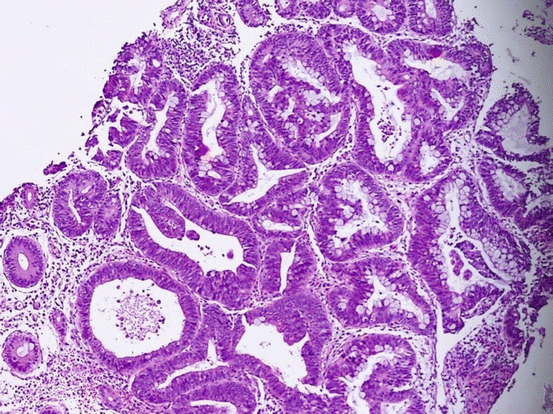
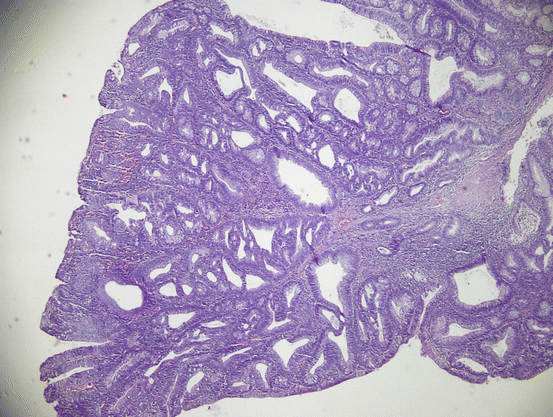
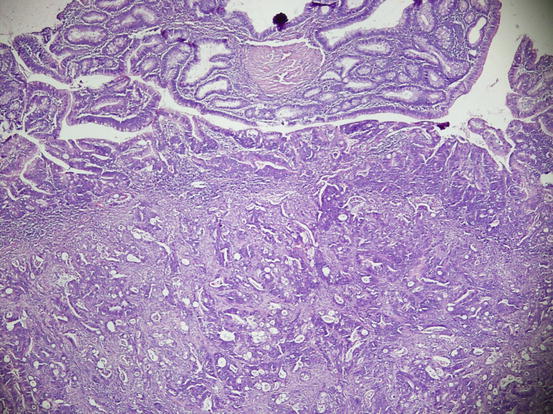
Fig. 11.14
Back-to-back glands with cribriform architecture and high-grade dysplasia and invasion into the lamina propria (HE ×100)
Fig. 11.15
Intramucosal adenocarcinoma due to invasion into muscularis mucosa in TA (HE ×40)
Fig. 11.16
Invasive carcinoma occurs with penetration beyond muscularis mucosa and invasion to the submucosal peduncle in TVA (HE ×40)
Natural History and Treatment
Half or two-third of adenomatous polyps, which are clinically the most common and important polyps, have a high risk for CRC [169]. The rate of recurrence was reported to be about 15 % in adenomas [153]. Patient’s age, histologic type, size and number of the polyps are important parameters for progression [150, 169]. When compared to individuals with one polyp, there is a twofold increased risk in those with three– to four polyps, and a fourfold increased risk in those with five or more polyps for carcinoma development [150]. As previously reported, advanced adenomas represented a big risk for development of invasive CRC [89, 130, 169–171]. Prognosis of polyps depending on their diameter remains controversial. While some argue that the diameter has no impact on local recurrence and lymphoid node metastasis in patients under surveillance following the treatment [162, 177], there are some other studies that have suggested that the risk for malignancy increases with increasing size of the polyp [178]. Another study that compared polyps smaller than 1 cm in size with diminutive (<0.6 cm) polyps reported that polyps smaller than 1 cm were more neoplastic than diminutive polyps, and the risk for carcinoma was increased in the patients older than 60 years, left colon polyps, and in males [179]. Although most of the adenomas are located distal to the splenic flexure, advanced neoplasia or adenoma has been shown in proximal to the splenic flexure in these patients, and examination of the entire colon is needed since it may show an advanced neoplasia without presence of any distal polyp [147, 163, 180, 181]. Thus, colonoscopy is an optimal screening method over all other modalities such as anoscopy, sigmoidoscopy, occult blood testing in stool, and barium enema [147]. Screening programs with colonoscopy to prevent colorectal adenocarcinoma are based on distribution of the adenomas in the colon, age of the patient, and family history [98, 130]. According to current consensus, colonoscopy screening starts at age 50 or 10 years earlier than the age of a first-degree relative who developed cancer [148, 174]. Based on the number of adenomas, on type and diameter of polyps during the first colonoscopy, and on family history, it is grouped as low-, moderate-, and high-risk adenoma, and it is repeated every 1–10 year depending on the risk of the adenoma removed during colonoscopy [130, 149, 168, 182]. Surveillance chart recommended by several studies is summarized in Table 11.2 [130, 147, 168, 182].
Table 11.2




Colonoscopy chart for adenomas
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
