Persistent AR signaling
Stress response pathways
Signaling pathways
Developmental pathways
AR, N-Terminal AR, co-factors LSD-1 and BF3
Hsp27, CLU, autophagy, Hsp90, p38 MAPK
PI3K/Akt, MAPK, JNK, NF-κB, CCL2, MET, src
EMT (N-Cad, Zeb 1,2, Twist, TGF-B, IL-6); WNT, Hedgehog (Shh), MAPK, IL8, Trop-2
Co-targeting Microtubules with Simultaneous Chemo-Hormonal Combinations
Docetaxel and cabazitaxel are the two taxanes approved for clinical use in CRPC [9, 10]. Docetaxel was the first approved non-hormonal therapy with a demonstrated survival benefit in CRPC. The TAX327 study showed that docetaxel every 3 weeks plus prednisone was superior to weekly mitoxantrone plus prednisone [10]. Median survival in the q3 weekly docetaxel plus prednisone arm was 19.2 versus 16.3 months in the mitoxantrone/prednisone arm [9]. The TROPIC study established the role of cabazitaxel as second-line therapy in CRPC, randomizing men with progressive disease during or after docetaxel to cabazitaxel + prednisone versus mitoxantrone + prednisone [11], and demonstrated 2.4 months improved overall survival with cabazitaxel.
Taxanes function by stabilizing the dynamic polymerization of microtubules. The ability of microtubules to assemble and disassemble is critical for mitosis and thus targeting microtubules preferentially in rapidly dividing cancer cells. Resistance to taxanes may be mediated through overexpression of the multi-drug-resistant P-glycoprotein efflux pump [12], mutations in the microtubule binding sites, and mutations in microtubule-associated proteins giving greater stability to cellular microtubule assembly [13, 14].
Taxanes also affects AR signaling through its alteration of microtubules-associated AR cellular transport and nuclear translocation [15, 16], raising speculation about cross-resistance with AR pathway inhibitors and questions of simultaneous verses sequencing of combinatorial chemo-hormonal regimens. Eigl et al. [17] demonstrated that simultaneous combined taxane-based chemotherapy plus androgen ablation is significantly more effective than the sequential administration of these treatments in the castrate sensitive Shionogi and LNCaP tumor models. Interestingly, a lack of response to castration was observed after initial paclitaxel therapy while gene expression studies confirmed up-regulated of several genes known to play a role in castrate resistance in response to paclitaxel exposure. These findings illustrate how stress-induced gene expression changes after chemotherapy or castration can confer cross-treatment resistance and provide preclinical proof-of-principle for ongoing clinical trials addressing the role and timing of systemic therapies in prostate cancer.
ECOG3805 (CHAARTED) compared standard sequence ADT followed by docetaxel upon castration resistance versus docetaxel given at the start of ADT in 790 men with metastatic castrate sensitive prostate cancer (http://clinicaltrials.gov/show/NCT00309985). Men received either ADT alone or ADT with the chemotherapy drug docetaxel every three weeks over a period of 18 weeks. Men who received simultaneous combinatorial chemo-hormonal lived longer than patients who received ADT alone with 3-year overall survival of 69.0 versus 52.5 %, respectively (nih.gov/news/health/dec2013/nci-05.htm). Patients with a high extent of metastatic disease accounted for most of the benefit in the overall survival from docetaxel plus ADT (3-year survival rates of 63.4 versus 43.9 % for ADT alone). While further follow-up is needed in order to define the effect of this treatment combination on patients with less extensive metastatic disease, this study provides the first example of clinical efficacy using combinatorial therapies in advance prostate cancer and supports the early addition of docetaxel to ADT rather than waiting until CRPC.
Reciprocal Cross-Talk Activation of AKT and MAPK
CRPC is a complex process by which cells survive and proliferate in low circulating androgen. This in part involves the reactivation of the androgen receptor (AR) axis [18], by pro-survival genes and alternative mitogenic growth factor pathways [19–22] including the phosphoinositide 3-kinase (PI3K)/AKT pathway. Indeed, the AR (via amplification) and PI3K/AKT (via Pten loss) pathways are the two most frequently activated signaling pathways in prostate cancer. While AR inhibitors confer clinical responses in most patients, PI3K inhibitors rarely induce tumor regression in preclinical models. Indeed, monotherapy with AR or AKT inhibitors result in reciprocal cross-talk activation that helps emergence of acquired resistance [23–26]. For example, Carver et al. [23] showed that these pathways regulate each other via reciprocal negative feedback, such that inhibition of one activates the other. Inhibition of the PI3K pathway restored AR signaling in PTEN-deficient prostate cells in part through relief of negative feedback to HER kinases; conversely, blockade of AR relieves feedback inhibition of AKT through reduced levels of FKBP5 impairing the stability of the phosphatase PHLPP. Hence while tumor cells could adapt and survive when either single pathway is inhibited pharmacologically, combined inhibition of PI3K/AKT and AR signaling using the PI3K/mTOR inhibitor BEZ235 and the AR antagonist ENZ significantly delayed CRPC progression in the LNCaP model [23].
Thomas et al. [27] reported that monotherapy with the AKT-inhibitor AZD5363 increased AR transcriptional activity and AR-dependent genes such as PSA and NKX3.1 expression. These effects were overcome by the combination of AZD5363 with bicalutamide resulting in synergistic inhibition of cell proliferation and induction of apoptosis in vitro, and prolongation of tumor growth inhibition and PSA stabilization in CRPC in vivo. These studies provide preclinical proof-of-concept that combination co-targeting of an AKT inhibitor with an AR antagonist leads to prolonged delay of CRPC progression.
In addition to the AKT pathway, other signaling pathways have been identified that are induced by AR blockade-induced cross-talk activation (Fig. 16.1). For example, AR blockade leads to activation of MAPK pathway which is coordinated by a feed-forward loop involving p90rsk-mediated phospho-activation of YB-1 with subsequently induces the activation of survival pathway leading to the up-regulation of the molecular chaperone CLU, hence inducing treatment resistance [28]. Interestingly, targeting CLU using OGX-011 currently in phase III clinical trials in combination with ENZ not only delay the resistance to ENZ but also inhibit both AKT and MAPK pathways and further provide a preclinical proof-of-principle of co-targeting AR pathway in combination with OGX-011 [28].
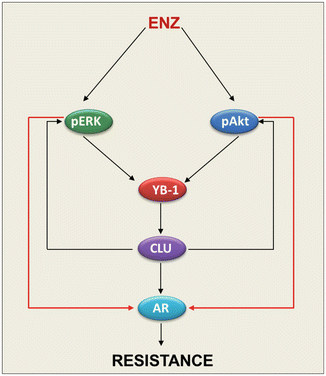
Fig. 16.1
ENZ induces treatment resistance via feed-forward mechanisms involving MAPK and AKT pathways and molecular chaperone CLU
Molecular Chaperones and the Stress Response
In cancer, stress is a driving force behind evolution (oncogenesis) and adaptation (acquired treatment resistance). During transformation, tumor cells undergo drastic shifts in their intracellular and extracellular milieu, frequently exposed to stress microenvironments that include hypoxia, acidosis, nutrient deprivation, and immune attacks from the host. To survive and prosper, like organisms living in the wild, tumor cells must be able to adapt to a variety of stress conditions. Many anti-cancer treatments induce stress responses that inhibit apoptosis and promote emergence of an acquired treatment-resistant phenotype. The heat shock response, for instance, is a highly conserved protective mechanism in eukaryotic cells associated with survival, thermo-tolerance, and oncogenic transformation [29]. Molecular chaperones play key roles in these stress responses, facilitating treatment resistance by regulating protein homeostasis (proteostasis) as well as many signaling and transcriptional survival networks. Chaperones play central roles in endoplasmic reticular (ER) stress [30–32] and the unfolded protein response (UPR), tailored to re-establish proteostasis by inhibiting translation, increasing chaperone expression, and promoting proteasome- and autophagy-mediated protein degradation. While these adaptive responses are cytoprotective, cell death can occur when ER stress and misfolded protein burden overwhelms the degradation capacity of the proteasome or autophagy [33–38]. Co-targeting stress-induced survival pathways regulating proteostasis, such as ERAD or ERAA, may better manipulate cancer cell sensitivity to therapy.
Molecular chaperones, including heat-shock proteins (Hsps), are key mediators of the stress response, acting as genetic buffer to stabilize and help cells cope at times of environmental stress. During cancer progression, Hsps increase Darwinian fitness of cells during transformation, progression, and treatment resistance [39]. Molecular chaperones bind misfolded proteins to facilitate substrate refolding or degradation, protecting cells against protein aggregation, and facilitating refolding or degradation. Chaperone expression is induced after many varied insults including hypoxia, heat shock, anoxia, glucose deprivation, free radicals, carcinogens, hormone therapy, and chemotherapy [40–43]. Indeed, expression of sHSPs is frequently up-regulated in many cancers and correlates with metastases, poor response to chemotherapy, and poor survival.
The transcriptional regulation of sHSPs is convoluted because of redundancy and feedback control, but several stress-related transcription factors are considered to be major inducers of HSPs. Heat shock factor 1 (HSF-1) is a highly conserved transcription factor that binds to consensus heat shock element (HSE) within the promoter regions of HSP genes [44]. Stress-induction of HSF-1 is a multi-step process that involves constitutive expression of HSF-1 monomer, stress-induced HSF-1 serine phosphorylation, followed by its trimerization, nuclear translocation with increased transcription [45]. HSF1 activates the transcription not only for sHSPs but also other molecular chaperones that associate with HSF1 to initiate a negative-feedback loop and inhibit HSF1 transcriptional activity [46]. Deletion of Hsf1 gene in mammalian cells does not alter normal basal expression of HSPs but abrogates stress-induced expression of HSPs [47], suggesting that other transcription factors are involved in the basal expression of HSPs. In mice, HSF-1 deficiency inhibits spontaneous tumor formation initiated by dominant negative mutation of p53 and chemical induced skin carcinogenesis associated with activating mutations of H-Ras proto-oncogene [29]. While HSF-1 is not a classic oncogene capable of driving transformation, it does coordinate a broad of network of cellular functions that supports tumorigenesis, highlighting HSF-1 as key regulator of the stress response that is usurped during oncogenesis to promote adaptation and malignant progression.
This section will review roles for Hsp27 and CLU in stress response and acquired treatment resistance, and their current status as therapeutic target in CRPC.
Hsp27
Hsp27 (HspB1) is transcriptionally regulated by HSF-1 and several studies suggest that the specific transactivation of Hsp27 is stimulus dependent [48–50]. The transcriptional regulation of Hsp27 appears to be regulated by many factors influenced by cell type and cell context. Additionally, Hsp27 can be phosphorylated on three serines (Ser15, ser78, and Ser82) and on Threonine (Thr143). Hsp27 phosphorylation is a reversible process catalyzed by a large number of kinases including MAPKAP kinases 2 and 3, p90Rsk, PKC, PKD, and PKG. Hsp27 becomes phosphorylated in response to a variety of stresses, including oxidative stress, inflammatory cytokines like tumor necrosis factor-α(TNF-α) Interleukin-1β, and transforming growth factor (TGF-β), and mitogens like insulin like growth factor (IGF-1) [51], and steroid hormones [52]. Hsp27 phosphorylation plays an important role in cytoskeleton organization and cell migration. Hence, Hsp27 phosphorylation is required for TGF-β induced metalloproteinase-2 activation and invasion in breast cancer cells and that inhibiting Hsp27 phosphorylation resulted in inhibition of tumor cell migration and invasion [53]. Hsp27 phosphorylation is associated with many malignancies and correlates with p90Rsk in prostate cancer [51] as well as progression to castrate resistance [51].
(a)
Hsp27 regulates cell survival. Hsp27 is highly and uniformly expressed in treatment-resistant cancers including CRPC [54, 55]. Its expression is induced by hormonal withdrawal and/or chemotherapy, and inhibits treatment-induced apoptosis through multiple mechanisms [56–60]. Hsp27 interacts with and inhibits components of both stress- and receptor-induced apoptotic pathways. It prevents activation of caspases by sequestering cytoplasmic Cytochrome C [61]; interacts with and inhibits Caspase-3 activation; and stabilizes actin microfilaments [62] by binding to F-actin preventing disruption of the cytoskeleton [63]. Hsp27 also regulates Akt and inhibits Bax activation [64]. Hsp27 inhibits apoptosis induced by etoposide or TNF-α in different cancer cell lines by increasing IκBα ubiquitination and degradation, leading to the activation of NF-κB [65]. In AR positive LNCaP prostate cancer cells, androgen stimulates Hsp27 phosphorylation via p38 kinase, which displaces Hsp90 as an AR chaperone and then shuttles AR to the nucleus to facilitate binding to androgen response elements and enhance AR transcription activity and cell survival [52].
After androgen withdrawal, several growth factor pathways play an important role in PCa cell survival, including IGF-1 and IL-6. IGF-1 stimulates Hsp27 phosphorylation in a dose and time dependent manner; Hsp27 is a novel p90Rsk substrate and downstream effector of Erk, increasing IGF-1 induced phosphorylation of Erk, p90Rsk, and Akt [51]. Conversely, Hsp27 knockdown abrogates IGF-1-induced phosphorylation of Erk, p90Rsk and Akt, thereby destabilizing Bad/14-3-3 complexes and increasing apoptotic rates [51]. Hsp27 regulates activity of oncogenic signaling pathways, such as STAT3 [56, 66, 67] which controls expression of cell survival genes and pathways associated with treatment resistance. Hsp27 has a “switch” role in regulating PEA-15 activity to enhance cell proliferation and suppress Fas-induced cell death [68]. Hsp27 enhances Akt activation which, in turn, phosphorylates PEA-15 at Ser-116, switching the binding specificity of PEA-15 from ERK to FADD. The dissociation of PEA-15 from ERK alleviates the cytoplasmic sequestration of ERK, allowing its translocation into the nucleus where it mediates its mitogenic effects. Upon phosphorylation of PEA-15 at Ser-116, PEA-15 binds to FADD and thereby inhibits Fas-induced apoptosis. These findings suggest that patients with tumors harboring abnormalities in PTEN function may clinically respond more favorably to Hsp27 inhibitors [68], thus identifying a population more likely to benefit from Hsp27 inhibition therapy.
(b)
Hsp27 regulates EMT and metastasis. In addition to its role in cell survival, Hsp27 drives epithelial–mesenchymal-transition (EMT) in PCa, whereas its attenuation reverses EMT and decreases cell migration, invasion, and matrix metalloproteinase activity. Mechanistically, silencing Hsp27 decreased IL-6-dependent STAT3 phosphorylation, nuclear translocation, and STAT3 binding to the Twist promoter, suggesting that Hsp27 is required for IL-6-mediated EMT via modulation of STAT3/Twist signaling. A correlation between Hsp27 and Twist has been observed in patients with prostate cancer, with Hsp27 and Twist expression each elevated in high-grade prostate cancer tumors. Hsp27 inhibition by OGX-427, an antisense therapy currently in phase II trials, reduced tumor metastasis in a murine model of prostate cancer. More importantly, OGX-427 treatment decreased the number of circulating tumor cells (CTCs) in patients with metastatic castration-resistant prostate cancer in a phase I clinical trial. Overall, this study defines Hsp27 as a critical regulator of IL-6-dependent and IL-6-independent EMT, validating this chaperone as a therapeutic target to treat metastatic prostate cancer. Moreover, it regulates actin rearrangement, cytoskeleton organization and cell migration [53] and thereby enhances cell migration and invasion via modulation of Fak-dependent actin organization and STAT3-dependent MMP-2 expression [69]. Knockdown of Hsp27 inhibits VEGF-induced cell migration [70] and abrogates TGF-β induced MMP-2 as well as cell invasion in human prostate cancer cell lines [71, 72].
(c)
Hsp27 as a prognostic marker. Clinically, Hsp27 is highly expressed in many cancers including prostate [54, 55] and others [73] and is associated with aggressive tumor behavior, metastasis and poor prognosis [60, 74]. In prostate cancer, expression of Hsp27 in diagnostic biopsy predicts poor clinical outcome [75] and correlates with CRPC progression [56]. Hsp27 expression and phosphorylation correlate with CRPC progression [51, 56]. Hsp27 expression increases shortly after treatment with androgen ablation and becomes highly uniformly expressed in CRPC [51, 54, 56, 74]. Hsp27 expression at diagnosis predicts poor clinical outcome independent of ETS-gene rearrangement [75].
(d)
Hsp27 as a therapeutic target. As a stress-activated chaperone, Hsp27 expression is induced by hormone and chemotherapy and inhibits treatment-induced apoptosis through multiple mechanisms [51, 56–60]. Consistent with its multiple cytoprotective functions, overexpression of Hsp27 renders human prostate LNCaP tumors resistant to paclitaxel, enhances tumor growth, and confers resistance post castration [56, 76]. Hsp27 interacts with factors involved in oncogenic signaling pathways and CRPC progression such as STAT3, IGF-1, AR, and eIF4E [52, 56, 66, 76]. For example, Hsp27 binds to AR and displaces Hsp90 as the predominant chaperone after androgen treatment, shuttles the AR to the nucleus, and facilitates AR binding to the androgen response element in AR-regulated genes [52]. Additionally, after castration therapy, increased Hsp27 confers resistance by activating STAT3 [56], and by stabilizing eIF4E e- to inhibit its stress-induced ubiquitination and proteasomal degradation to enhance survival [76].
OGX-427 (OncoGeneX Pharmaceuticals) is a second-generation 2′-methoxyethyl phosphorothioate ASO targeting Hsp27 that has a tissue half-life of >7 days. Rocchi et al. first reported ASO-induced Hsp27 knockdown induced apoptosis with single agent tumor growth inhibition as well as enhanced hormone- and chemo-therapy activity when used in combination [74, 77]. Hsp27 inhibition with OGX-427 demonstrated anti-cancer activity in vitro and in vivo [52, 56, 74, 78], inducing AR [52] and eIF4E [76] proteasomal degradation with decreased AR transactivation and PSA expression in vivo [52]. Hsp27 ASO induces apoptosis and delays prostate tumor progression [74] and chemo-sensitizes bladder, prostate, ovarian, and uterine cancer to paclitaxel [74, 79, 80], and inhibits metastasis dissemination in murine model of prostate cancer [67]. Since Hsp27 functions as a regulatory “hub” in multiple adaptive survival signaling and transcriptional pathways, it is an attractive therapeutic target; Hsp27 inhibition may simultaneously suppress many pathways implicated in cancer progression and resistance to hormone- and chemo-therapies.
OGX-427 has completed single agent and docetaxel-combination dose escalation phase I trials in prostate, bladder, breast, and lung cancer. OGX-427 is well tolerated at 800 and 1,000 mg dose in combination with docetaxel. Reduction in tumor markers was observed in patients with prostate (PSA) and ovarian (CA-125) cancer. Decline of 50 % or greater in both total and Hsp27+ CTCs was observed in over half of the patients [67]. A randomized phase II study of OGX-427 plus prednisone versus prednisone alone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (CRPC) has also been completed. Patients with CRPC and no prior treatment were enrolled 1:1 to receive prednisone 5 mg PO BID alone or with OGX-427 600 mg IV × 3 loading doses in week 1 followed weekly 2-h infusions on a 4 week cycle. Preliminary results indicated that 82 % of patients treated with OGX + prednisone had a PSA decline (45 % with >30 % decline) and 18 % a PSA increase compared to 50 % of patients treated with prednisone have had a PSA decline (20 % with >30 % decline) and 50 % a PSA increase [81]. CTC conversion from ≥5 to <5/7.5 ml occurred in 50 % of patients treated with OGX + prednisone and 20 % of patients treated with prednisone alone. These results confirm, for the first time, single agent activity for an Hsp27 inhibitor in cancer, and phase II combination studies are ongoing in CRPC, lung, pancreas, and bladder cancer.
Clusterin
Secretory CLU (CLU) is a multifunctional, stress-induced, ATP-independent molecular chaperone containing amphipathic and coiled-coil helices in addition to large intrinsic disordered regions. These properties of CLU resemble survival chaperones associated with tissue injury and pathology like acute phase protein haptoglobin [82] and small Hsp’s [83]. Indeed, CLU is involved in many biological processes ranging from mammary and prostate gland involution to amyloidosis and neurodegenerative disease, as well as cancer progression and treatment resistance [84]. Promoter sequences of CLU gene are conserved during evolution and include stress-associated sites like activator-protein-1 (AP-1), AP-2, SP-1, HSE (heat shock element) recognized by HSF-1/HSF-2 heterocomplexes, and CRE (cAMP response element) [85]. Additionally, there are glucocorticoid response element (GRE) [86–88], androgen response element (ARE) [89], and Y-box binding protein (YB-1) sites [90]. CLU expression increases downstream of survival signaling pathways and in response to ER-stress. CLU is up-regulated downstream IGF-1 via Src-Mek-Erk-EGR-1 [91] and cytokines via Jak/STAT1 [92] and downstream of ER stress inducer including paclitaxel via YB-1. YB-1 directly binds to CLU promoter regions to transcriptionally regulate clusterin expression. In response to endoplasmic reticulum stress inducers, including paclitaxel, YB-1 is translocated to the nucleus to transactivate CLU. YB-1 transactivation of CLU in response to stress is a critical mediator of paclitaxel resistance in prostate cancer [90].
CLU Enhances Cell Survival Under Stress Conditions
sCLU functions to protect cancer cells from many varied therapeutic stressors that induce apoptosis, including androgen or estrogen withdrawal, radiation, chemotherapy, and biologic agents [93]. CLU is regulated by HSF1 [85], and functions like small HSPs to chaperone and stabilize conformations of proteins at times of cell stress, potently inhibiting stress-induced protein precipitation by binding to exposed regions of hydrophobicity on non-native proteins to form soluble, high molecular mass complexes [94–96]. CLU inhibits ER stress, retro-translocating from the ER to the cytosol to inhibit aggregation of intracellular proteins and prevent apoptosis [97]. Interestingly, CLU is the most abundant protein associated with β-amyloid deposits in Alzheimers, likely related to its role in inhibiting protein aggregation [94]. Collectively, the preceding indicates sCLU plays an important role in protein homeostasis (proteostasis) via unfolded protein and ER stress responses.
CLU inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizes cytosolic Ku70-Bax protein complex to inhibit Bax activation [98], interacting with conformationally altered Bax to inhibit apoptosis in response to chemotherapeutic drugs [99]. In addition, CLU increases Akt phosphorylation levels and cell survival rates [100] and promotes prostate cancer cell survival by increasing NF-κB nuclear transactivation, acting as a ubiquitin-binding protein that enhances COMMD1 and I-kB proteasomal degradation via interaction with E3 ligase family members [101]. Experimental and clinical studies associate CLU with development treatment resistance, where CLU suppresses treatment-induced cell death in response to androgen withdrawal, chemotherapy, or radiation [102–104].
CLU Correlates with Adverse Prognosis
CLU is expressed in many human cancers, including breast, lung, bladder, kidney, colorectal, and prostate [105–109]. In prostate, CLU was originally cloned as “testosterone-repressed prostate message 2” (TRPM-2) [110] from regressing rat prostate, but was later defined as a stress-activated and apoptosis-associated, rather than an androgen-repressed, gene [102]. CLU expression correlates with loss of the tumor suppressor gene Nkx3.1 during the initial stages of prostate tumorigenesis in Nkx3.1 knockout mice [111]. High levels of CLU expression associate with migration, invasion and metastasis, increasing Smad2/3 stability and enhancing TGF-β-mediated Smad transcriptional activity [112]. In contrast, CLU silencing induces mesenchymal–epithelial-transition via inhibition of Slug [113].
CLU levels are low in low-grade cancers, but increase with higher Gleason score [109]. Levels of sCLU increased several fold after androgen ablation, consistent with its stress-activated, cytoprotective response to anti-cancer treatment [104]. Biochemical recurrence-free survival in patients with strong CLU expression in prostatectomy specimens was lower than those with weak CLU expression [114]. Plasma levels of sCLU were significantly higher in patients with high-grade prostate cancer with extra-capsular extension compared to organ-confined tumors [115]. These data correlate CLU with higher grade, post treatment stress, and/or poor outcome in many cancers.
CLU as Therapeutic Target
CLU levels are high in CRPC [102, 103] and after estrogen withdrawal in breast cancer cells [116–119], cisplatin [120, 121], doxorubicin [122, 123], Herceptin [124], Hsp90 inhibitors [125], and HDAC inhibitors [126]. In keeping with its cytoprotective function, CLU inhibition enhances cytotoxicity of hormone- radiation-, and chemo-therapies [119, 127, 128]. The antisense inhibitor, OGX-011 (custirsen), is a second-generation ASO with a long tissue half-life of ~7 days that potently suppresses CLU levels in vitro and in vivo. OGX-011 improved the efficacy of many varied anti-cancer therapies by suppressing treatment-induced CLU and the stress response [125] preclinical activity in many xenograft models of cancer [128–131]. CLU ASO sensitizes bladder cancer to cisplatin [121], prostate to paclitaxel [103], and prostate to hormone therapy [102] where it delays CRPC progression [121, 102, 125, 132].
The first-in-human phase I study with OGX-011 used a novel neoadjuvant design to identify effective biologic dosing of OGX-011 to inhibit sCLU expression in human cancer [133]. Neoadjuvant androgen deprivation was administered concurrently. At doses of 320 mg and higher, concentrations of full-length OGX-011 were achieved that were associated with preclinical activity. OGX-011 produced statistically significant, dose-dependent >90 % knockdown of CLU in normal and tumor tissue and identified 640 mg as the optimal biologic dose for Phase II trials. Plasma pharmacokinetic parameters have been similar across phase I studies including when OGX-011 was combined with chemotherapy and decreases in serum sCLU have been consistently observed [134, 135]. A phase II trial of 85 patients with non-small cell lung cancer (NSCLC) treated with combined OGX-011 and gemcitabine-cisplatin chemotherapy [135, 136] reported an objective response rate of 23 % and median overall survival of 383 days with 58 % surviving >1 year.
Related posts:
 Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
 Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
 Phase I–II Targeted Treatments
Phase I–II Targeted Treatments
 Epigenetics in Castration Resistant Prostate Cancer
Epigenetics in Castration Resistant Prostate Cancer
 Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


