Fig. 16.1
Structures of deoxyadenosine, cladribine, fludarabine, and clofarabine
The basis of selectivity with antiviral agents usually involves selective activation by an enzyme expressed by the virus or selective inhibition of an enzyme important to the synthesis of the viral genetic material. With anticancer analogs, the basis for selectivity is much less clear because the enzymes involved in activation and activity in cancer cells are identical to those found in normal cells. However, the antitumor selectivity of nucleoside analogs can be attributed to enhanced metabolism in cancer cells or inhibition of DNA synthesis or function. The goal of cancer drugs is to prevent replication of the cancer cell because it is this replication that leads to the death of the patient. Since most host cells in humans are not replicating and therefore are not sensitive to agents that inhibit DNA synthesis, inhibition of DNA synthesis in cancer cells affords modest selectivity. However, some tissues have cells with rapid turnover (bone marrow, gastrointestinal tract, and hair follicles), and toxicity in these tissues due to the inhibition of DNA synthesis limits the amount of drug that can be administered.
Because clofarabine is an analog of 2′-deoxyadenosine (dAdo), the metabolism of dAdo in human cells, summarized in Fig. 16.2, introduces the enzymes that are important in the activation and antitumor activity of clofarabine. Adenosine deaminase is expressed in the plasma; therefore, the primary metabolic route for dAdo that is injected into animals is deamination by adenosine deaminase to deoxyinosine, which is rapidly cleaved by purine nucleoside phosphorylase (PNP) to hypoxanthine. Hypoxanthine can be reused by hypoxanthine/guanine phosphoribosyl transferase (HGPRT) for the generation of purine nucleotides, although it is more likely to be converted by xanthine oxidase to xanthine and uric acid, which is eventually excreted. Adenine-containing nucleosides are extremely poor substrates for mammalian PNP [3]; therefore, little, if any, dAdo that is administered intravenously is cleaved to adenine in mammalian systems. However, dAdo is a good substrate for bacterial PNP [4] and would be readily cleaved in the gastrointestinal tract. A small amount of dAdo administered to animals is phosphorylated to dAMP, primarily by deoxycytidine kinase (dCyd kinase), which is further phosphorylated to dADP and dATP. dATP is the primary acid-soluble intracellular metabolite of dAdo, and it is used as a substrate for the synthesis of DNA. An important enzyme in the generation of deoxynucleotides for DNA synthesis is ribonucleotide reductase. The activity of this enzyme is controlled by the natural NTPs and dNTPs to maintain the proper balance of nucleotides for DNA synthesis. As can be seen in Fig. 16.2, dATP represses ribonucleotide reductase activity, whereas ATP stimulates its activity.
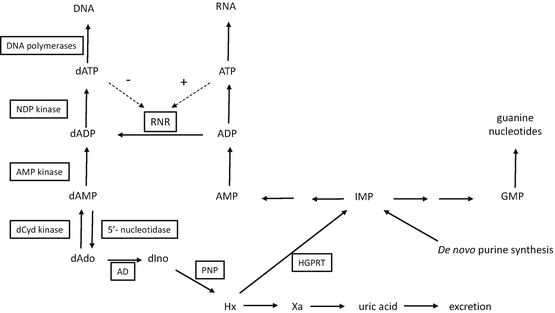
Fig. 16.2
Schema depicting the metabolism of deoxyadenosine. Enzymes are boxed in rectangles. Abbreviations: NDP nucleoside diphosphate, dCyd 2′-deoxycytidine, dAdo 2′-deoxyadenosine, dIno 2′-deoxyinosine, Hx hypoxanthine, Xa xanthine, AD adenosine deaminase, PNP purine nucleoside phosphorylase, HGPRT hypoxanthine-guanine phosphoribosyl transferase, RNR ribonucleotide reductase, IMP inosine 5′-monophosphate, GMP guanosine 5′-monophosphate, AMP adenosine 5′-monophosphate
16.2 Rationale for the Synthesis of Clofarabine
Numerous enzymes are involved in the activation and activity of nucleoside analogs, which is a primary reason why drug discovery within this class of compounds is a very empirical process. It is difficult, if not impossible, to design a compound that will behave as needed with each of the enzymes that must be involved in its activity. Therefore, the rational drug discovery strategy with these molecules is to generate numerous analogs that are based on a thorough understanding of the biological activity of the many synthesized compounds and then evaluate them in appropriate model systems to identify the agents with the desired activities.
A thorough explanation of the rationale for the synthesis of clofarabine can be found in a review by Secrist et al. [5]. In brief, nucleosides with halogens at the two position of the adenine base are known to be very poor substrates for adenosine deaminase [6] and therefore are known to be stable in plasma. These compounds (2-halo-2′-deoxyadenosine analogs) potently inhibit tumor cell growth. In addition, 2′-flouro nucleosides have been synthesized and are known to have biological activity [7]. 2′-Halogens on nucleoside analogs are also resistant to cleavage by acid and purine nucleoside phosphorylases, another feature that would contribute to their in vivo stability. Therefore, in a search for new anticancer agents, numerous 2′-halo (F, Cl, or Br) and 2-halo (F, Cl, or Br) dAdo analogs have been synthesized and evaluated for biological activity in both in vitro and in vivo tumor models [8]. Clofarabine was the most potent compound in in vitro cell-killing assays and demonstrated excellent activity against numerous human tumor xenografts in mice [9, 10].
16.3 Transport
Human cells will salvage natural nucleosides, if available, for nucleic acid synthesis in place of de novo synthesis, and cells express numerous transporters to aid the entry of these compounds. Both equilibrative transporters (hENT1, hENT2, and hENT3) and concentrative transporters (hCNT1, hCNT2, and hCNT3) are expressed on most human cells [11]. Because of the abundance of these transporters, natural nucleosides in the cell culture will equilibrate across the cell membrane in just a few seconds. To be active, all nucleoside analogs (with the possible exceptions of deoxycoformycin and forodesine) must be able to cross cellular membranes to interact with their cellular targets. Most nucleoside analogs that are useful in the treatment of various diseases are recognized by these transporters; therefore, they also equilibrate across cell membranes very quickly. Precise transport studies have been conducted with clofarabine, and it has been shown that it is transported into cells by both human equilibrative and concentrative nucleoside transporters [12]. Cells producing hENT1 exhibited the highest uptake of clofarabine (0.14 pmole/μl), followed by hCNT2 (0.025 pmole/μl), whereas uptake in cells producing hENT2 or hCNT1 was not higher than that in cells that were nucleoside transport deficient. In oocytes expressing recombinant transporters, the efficiency of transport by hCNT3 (1.3) was much greater than that of hENT1, hENT2, or hCNT2 (0.11, 0.20, or 0.15). Although not entirely consistent, these results indicate that clofarabine can enter cells by all known transporters except hCNT1, which is selective for pyrimidine nucleosides. In addition, at high concentrations, diffusion of clofarabine directly through the membrane is also possible [12].
Comparative studies have shown [12] that cladribine had the highest efficiency of transport with hENT1, with a Vmax/Km value of 1.8 versus 0.7 for clofarabine and 0.8 for fludarabine (9-β-D-[arabinofuranosyl]-2-F-adenine). The uptake and flux of clofarabine, cladribine, and fludarabine were very similar in human renal proximal tubule cells, which express equilibrative hENT1, hENT2, and hCNT3 [13]. These results indicate that although there are some differences in transport, all of these compounds are readily transported across human membranes.
Some studies have evaluated the importance of ABCG2 to the activity of clofarabine. This ABC transporter is known as the breast cancer resistance protein and transports various metabolites, including nucleoside monophosphates, out of cells. Cells transfected with DNA constructs, resulting in overexpression of human or mouse ABCG2, had significantly reduced transport of both clofarabine and cladribine metabolites [14]. These results were correlated with decreased levels of cellular metabolites of cladribine in ABCG2-expressing cells, which were much less sensitive to clofarabine and cladribine. In these studies, the ABCG2 transporter transported both cladribine monophosphate and cladribine itself. The results of a study by Nagai et al. [15] supported these conclusions and showed that ABCG2 primarily transports clofarabine. Their results indicate that the metabolism of clofarabine and its cytotoxicity are strongly determined by the interplay between dCyd kinase and ABCG2, and they suggest that inhibition of ABCG2 will improve antitumor activity in tumors in which dCyd kinase levels are low. The role of ABC transporters in nucleoside analog metabolism and activity has recently been reviewed [16].
16.4 Metabolism
The metabolism of clofarabine is schematically presented in Fig. 16.3. Once inside the cell, clofarabine is converted to clofarabine triphosphate [17]. Xie and Plunkett [18] showed that at low concentrations, the primary intracellular metabolite was clofarabine monophosphate, which was approximately twice the level of clofarabine triphosphate. At high concentrations of clofarabine, the monophosphate and triphosphate levels were similar. Clofarabine diphosphate levels were very low, indicating that phosphorylation of the monophosphate of clofarabine is the rate-limiting step in its activation to the triphosphate. This characteristic is similar to that of cladribine [19] and indicates that the monophosphate kinase does not prefer molecules with substitutions at the two position of a purine nucleoside as large as a Cl atom. In the case of most other deoxynucleoside analogs used for the treatment of cancer, the nucleoside kinase is usually the rate-limiting enzyme in their activation, and the triphosphate usually accounts for more than 90% of the intracellular metabolites.
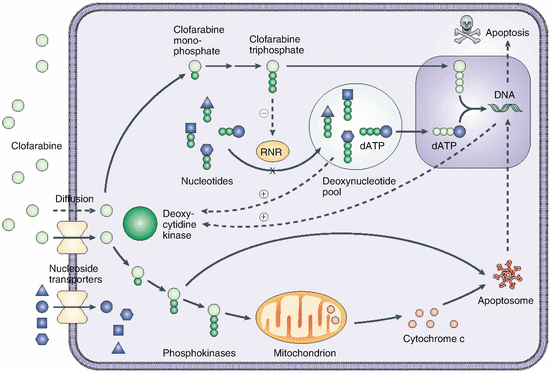
Fig. 16.3
Schematic of clofarabine’s mechanism of action. Clofarabine is transported into cells by both diffusion and facilitated diffusion. It is monophosphorylated by deoxycytidine kinase (dCK) and then serially phosphorylated by other kinases to form clofarabine triphosphate, the active moiety. The triphosphate acts to terminate DNA chain elongation and inhibit repair by incorporating it into the DNA chain through competitive inhibition of DNA polymerases. It also inhibits ribonucleotide reductase, with a reduction of dNTP pools, and induces apoptosis through direct and indirect actions on mitochondria by releasing cytochrome c and other proapoptotic factors (Obtained with permission from Bonate et al. [73])
Clofarabine triphosphate accumulates at high levels in CEM cells [17, 18]: treatment of cells with low concentrations of clofarabine (1–3 μM for 2–4 h) resulted in concentrations of clofarabine triphosphate that were 50–100 μM, which were similar to intracellular dATP concentrations in untreated cells. The accumulation of clofarabine triphosphate was dose dependent up to 10 μM. Similar intracellular concentrations of clofarabine triphosphate were achieved in all phases of the cell cycle (G1, S, and G2-M) [18]. Clinical studies have indicated that plasma clofarabine levels of 1.5 μM are achieved at the maximally tolerated dose of clofarabine in patients with acute leukemias after a 1-h infusion, and this treatment resulted in a clofarabine triphosphate concentration of approximately 19 μM in blast cells [20].
When clofarabine was removed from the medium of K562 and CEM cell cultures, clofarabine triphosphate had a half-life in cells of 1–3 h [18, 21]. However, in circulating leukemic cells, more than 50% of the clofarabine triphosphate was still present 24 h after clofarabine transfusion [20]. The metabolism of clofarabine and cladribine has been compared in ex vivo studies with CLL and AML cells that were obtained from patients [22]. In these studies, the half-life of clofarabine triphosphate was longer than that in CEM cells but similar to that of cladribine triphosphate (7.3 vs. 4.3 h, respectively). The enzymes involved in the degradation of nucleoside triphosphates in cells have not been studied in great detail. Triphosphate metabolites of nucleoside analogs do not readily cross cell membranes and are therefore trapped in the cells in which they were created, whereas nucleosides themselves will freely distribute across the cell membrane. Therefore, the antitumor activity of these agents can be extended well beyond the time that the drug circulates in the plasma because the active metabolite is maintained in tumor cells long after the drug has disappeared from the plasma. The long retention time of clofarabine triphosphate contributes to its lasting antitumor activity, even though the plasma half-life is fairly short [20, 23].
Cytoplasmic 5′-nucleotidase cN-II is one of the seven 5′-nucleotidases found in mammals. This enzyme catalyzes the removal of the 5′-phosphate from purine 5′-monophosphates and is involved in the maintenance of cellular nucleotide pools. Therefore, it can also degrade analogs of purine nucleoside monophosphates and could theoretically interfere with the activation of clofarabine. In cell culture experiments with CEM and RL cells, inhibition of this enzyme did not affect the cytotoxicity of clofarabine and had only modest effects on the induction of apoptosis [24], indicating that it has only a modest effect on the metabolism of clofarabine when clofarabine is constantly present in the cell culture medium. The results of these studies suggest that the monophosphate pool maintained by the dCyd kinase/5′-nucleotidase activity ratio in the presence of excess extracellular clofarabine is sufficient to maintain cytotoxic clofarabine triphosphate levels in the cell. However, it is possible that such inhibitors would have a more dramatic effect on metabolism after the removal of clofarabine from the external environment, which happens in vivo. As clofarabine triphosphate is degraded to the clofarabine monophosphate, inhibition of this enzyme could prevent further degradation, resulting in re-phosphorylation by the monophosphate kinase and an increase in the intracellular half-life of clofarabine triphosphate.
As expected for a dAdo analog, clofarabine was incorporated into DNA, but not RNA, indicating that it is a DNA-directed drug [18]. At low concentrations, clofarabine is mostly incorporated into internal positions in the DNA (>80%), suggesting that DNA synthesis is not inhibited by the incorporation of clofarabine monophosphate into the growing DNA strand. However, at high concentrations (1 or 10 μM), much more clofarabine is found at terminal positions in DNA. At 10 μM, 65% of the clofarabine in DNA is found at terminal positions, which suggests that DNA synthesis is inhibited at the site of incorporation.
The primary enzyme involved in the activation of clofarabine in tumor cells is dCyd kinase [17, 18, 22]. Clofarabine is a very good substrate for this enzyme, with Km and Vmax values that are similar to those of dCyd [21] but much better than those of dAdo (the catalytic efficiency of dAdo is only 0.3% that of dCyd). Clofarabine was modestly better substrate for dCyd kinase than was cladribine. Its catalytic efficiency was three to four times that of cladribine, with a Vmax that was ten times greater. The X-ray crystal structure of dCyd kinase with clofarabine in the active site has been determined [25]. The results of these studies indicate that the conformation of the enzyme/clofarabine complex was similar to structures of the pyrimidine-bound complexes and that the interactions between the 2-Cl group and its surrounding hydrophobic residues contribute to the high catalytic efficiency of dCyd kinase with clofarabine.
Decreased dCyd kinase activity has been shown to result in resistance to clofarabine in various cell culture systems [22, 26–29]. Decreased histone acetylation [28], but not methylation of the dCyd kinase gene [26, 28, 29], was associated with decreased dCyd kinase expression. In HL-60 cells, the decreased activation of clofarabine was associated with decreased expression of dCyd kinase, deoxyguanosine kinase, and hENT1, hENT2, and hCNT3 genes [30]. Studies have shown that there are genetic variations in the expression of dCyd kinase activity in patient populations and suggest that these variations are responsible for the variable rates of activation of clofarabine and other nucleoside analogs [31].
Treatment of cells with clofarabine and other nucleoside analogs can enhance dCyd kinase activity in many, but not all, cell lines [32–34]. Others have shown that inhibition of DNA synthesis can enhance dCyd kinase activity [35, 36] by phosphorylating dCyd kinase at serine 74 [35, 37, 38]. The enhancement of dCyd kinase activity by clofarabine can be exploited to improve antitumor activity by combining clofarabine with other nucleoside analogs that are activated by dCyd kinase [33, 34, 39–41].
Clofarabine is also a good substrate for deoxyguanosine kinase [42], an enzyme that is expressed in mitochondria. The catalytic efficiency of clofarabine with this enzyme is similar to that of both deoxyguanosine and cladribine, although both the Km and Vmax values for the two deoxyadenosine analogs are 10–20 times greater than are those for deoxyguanosine. The contribution of deoxyguanosine kinase to the phosphorylation of clofarabine in cells is low due to the much higher expression of dCyd kinase activity in most cell types [43, 44], but it could be important in cells that express low amounts of dCyd kinase. Resistance to clofarabine in CEM cells was correlated with decreased dCyd kinase activity but not with decreased deoxyguanosine kinase [22, 27].
16.5 Mechanisms of Action
The mechanism of action of clofarabine resembles that of other dAdo analogs and is schematically presented in Fig. 16.3 . Below, we describe the primary actions of this nucleoside analog.
16.5.1 Inhibition of DNA Synthesis
As with other anticancer nucleoside analogs, the primary activity of clofarabine that is responsible for its antitumor activity is the inhibition of DNA synthesis [17, 18, 21, 45]. RNA and protein synthesis are inhibited by clofarabine only at high concentrations. Clofarabine triphosphate also inhibited yeast poly(A) polymerase activity [46], but it is unclear how much the inhibition of this enzyme contributes to the antitumor activity of clofarabine. DNA synthesis is immediately inhibited in cells treated with clofarabine, and a 3-h incubation with 0.3 μM clofarabine resulted in prolonged inhibition [45], which did not recover to more than 20% of control values 72 h after removal of the drug. The immediate and long-lasting inhibition of DNA synthesis in cells treated with clofarabine strongly suggests that the enzymes that are critical for DNA replication, such as DNA polymerase and ribonucleotide reductase, are the primary targets for clofarabine and that disruption of DNA function as a result of the incorporation of clofarabine into daughter strands is of secondary importance to the activity of clofarabine.
16.5.1.1 Inhibition of Ribonucleotide Reductase
Incubation of cells with clofarabine results in significant decreases in intracellular deoxynucleotide pools [17, 18, 45], suggesting that clofarabine triphosphate is a potent inhibitor of ribonucleotide reductase, a critical enzyme that is involved in the de novo synthesis of deoxynucleotides [47]. This hypothesis was confirmed in experiments in which clofarabine triphosphate potently inhibited ribonucleotide reductase in cell-free extracts [17], and incubation of cells with clofarabine dramatically decreased the conversion of radiolabeled purines and pyrimidines to their respective deoxynucleotide triphosphates [45]. Clofarabine had the most impact on dCTP and dATP pools, a modest impact on dGTP pools, and no effect on dTTP pools [17, 45]. The concentration of clofarabine triphosphate that is required to inhibit ADP reduction from ribonucleotide reductase isolated from K562 cells by 50% is 65 nM, which is similar to that seen with cladribine triphosphate [17].
The activity of ribonucleotide reductase in cells is tightly controlled by the natural nucleotides to ensure that the cell has all of the deoxynucleotides needed for DNA synthesis in the correct concentrations. It is known that dATP is a potent regulator of ribonucleotide reductase activity and inhibits the reduction of ADP, UDP, and CDP [47]. Therefore, the results described above suggest that clofarabine triphosphate interacts with ribonucleotide reductase in the allosteric binding site as an analog of dATP. However, in studies with purified human ribonucleotide reductase, Aye and Stubbe [48] showed that both clofarabine diphosphate and clofarabine triphosphate were potent inhibitors of ribonucleotide reductase activity. They showed that clofarabine triphosphate is a rapid reversible inhibitor, with a Ki of 40 nM, and that clofarabine diphosphate is a slow-binding, reversible inhibitor, with a Ki of 17 nM. Their results indicated that clofarabine triphosphate bound to the allosteric binding site (A site) on the α subunit. The A site controls the rate of the reaction, and when dATP is bound, the enzyme is inactive. Clofarabine diphosphate bound to the substrate-binding site (C site). Although clofarabine diphosphate levels are much lower than clofarabine triphosphate levels in cells [18], the relatively long half-life of the inhibited state means that clofarabine diphosphate could still significantly contribute to the inhibition of ribonucleotide reductase in cells. Finally, inhibition of enzyme activity by either compound was associated with protein oligomerization that was more kinetically stable than were dATP-induced hexamers [48–50].
16.5.1.2 Inhibition of DNA Polymerization
Clearly, inhibition of ribonucleotide reductase is sufficient to result in the inhibition of DNA synthesis; however, clofarabine triphosphate is also a good substrate and inhibitor of DNA polymerases α and ε, two important enzymes that are involved in the replication of chromosomal DNA [17, 45]. Clofarabine triphosphate was found to be a potent inhibitor of the incorporation of dATP by DNA polymerase α, with a Ki value of approximately 1 μM (Km for dATP was 4 μM), but it was a weak inhibitor of DNA polymerase β (involved in DNA repair) and γ (involved in mitochondrial DNA synthesis). A nucleoside triphosphate analog can interact with a DNA polymerase in one of three ways: it can interfere with DNA synthesis without being used as a substrate; it can be used as a substrate but not interfere with continued DNA synthesis; or it can be used as a substrate and cause disruption of subsequent DNA chain elongation. The results of studies in intact cells with radiolabeled clofarabine indicate that it is incorporated into DNA in internal and terminal positions [18], but these studies did not indicate whether DNA synthesis was inhibited due to the incorporation. In addition, inhibition of dATP incorporation by isolated polymerases does not indicate that DNA synthesis is actually inhibited because it is possible that clofarabine triphosphate inhibited dATP incorporation by acting as an alternative substrate without interfering with subsequent elongation.
The results of DNA-sequencing studies supported those of the radiolabeled studies, showing that clofarabine triphosphate is used by DNA polymerase α [17], with Km and Vmax values that are similar to those of dATP (as well as cladribine triphosphate). These studies also showed that the ability of polymerase α to add new nucleotides after the incorporation of clofarabine triphosphate was significantly less than that after the incorporation of dATP. The Km of adding dGTP after the incorporation of clofarabine triphosphate was 100-fold greater than that after the incorporation of dAMP. The Vmax for the incorporation of dGTP was similar, regardless of whether dAMP or clofarabine monophosphate was incorporated previously. Interestingly, the incorporation of cladribine had only a modest effect on the subsequent use of dGTP as a substrate (only a threefold difference to that seen after the incorporation of dAdo). The effect of clofarabine incorporation on subsequent chain elongation was similar to that of fludarabine, an agent known to interfere with DNA synthesis. The effect of clofarabine triphosphate on DNA polymerase ε activity was similar to that seen with DNA polymerase α, except that it more effectively inhibited chain elongation by DNA polymerase ε [45]. This enhanced inhibition of chain elongation resulted in significant inhibition of DNA synthesis by this enzyme at clofarabine triphosphate concentrations that were only 3% those of dATP.
Under conditions in which control reactions extended all of the labeled DNA primer to the end of the template strand, DNA polymerase α was able to extend the DNA chain with clofarabine triphosphate in place of dATP across a single thymidine residue in the template strand, but it was not able to extend past two consecutive thymidine residues [17]. This result was very similar to that seen with fludarabine triphosphate but was different from that seen with cladribine triphosphate. DNA polymerase α was able to extend the DNA chain with cladribine triphosphate across two consecutive thymidine residues in the template strand, but it could not extend beyond three consecutive thymidine residues. These results indicate that clofarabine caused significant chain termination by DNA polymerase α, which is consistent with the immediate and lasting inhibition of DNA synthesis seen in cells treated with clofarabine [18].
These results seem to be in conflict with the results that indicate that at low concentrations, more than 80% of the clofarabine incorporated into DNA is incorporated into internal positions [18], which suggests that elongation of DNA is not inhibited by the incorporation of clofarabine into DNA. The above results indicate that DNA polymerases can easily extend the DNA chain across single thymidine residues with clofarabine triphosphate but that chain extension is dramatically inhibited by the incorporation of two successive clofarabine nucleotides. Since it is likely that single incorporations are more prevalent in DNA than are multiple incorporations, there would be many more internal clofarabine molecules in DNA, even though the inhibition of DNA synthesis due to chain termination is the primary action of clofarabine that is responsible for its antitumor activity. In addition, chain termination caused by two successive clofarabine incorporations would have one internal and one terminal, for a ratio of 50%. These results indicate that DNA replication is severely inhibited, even under circumstances in which most of the incorporated clofarabine is in internal positions in the DNA.
Clofarabine has also been shown to inhibit nucleotide excision DNA repair in chronic lymphocytic leukemia (CLL) lymphocytes isolated from patients that were induced by treatment with 4-hydroperoxycyclophosphamide [51]. Clofarabine was as potent as fludarabine in the inhibition of DNA repair in this model; however, clofarabine triphosphate demonstrated maximal inhibition at one-tenth the concentration of fludarabine triphosphate. Although not directly studied in this paper, the inhibition of nucleotide excision repair could be due to the inhibition of the DNA polymerases involved in the repair or to the inhibition of ribonucleotide reductase activity. In addition, inhibition of DNA repair could explain the activity of clofarabine against resting non-proliferating tumor cells [52].
Collectively, the results presented in the previous paragraphs indicate that DNA synthesis is inhibited in cells treated with clofarabine through two distinct but complementary actions: inhibition of ribonucleotide reductase and DNA polymerase activity. The potent inhibition of ribonucleotide reductase activity by clofarabine nucleotides enhances its inhibition of the replicative DNA polymerases (self-potentiation) by decreasing the intracellular concentration of the natural substrate, dATP, which competes with clofarabine triphosphate for use as a substrate by these enzymes (i.e., in the presence of ribonucleotide reductase inhibition, less clofarabine triphosphate is needed to inhibit DNA polymerase activity). Indeed, clofarabine combines the strong DNA-terminating activity of fludarabine (a weak inhibitor of ribonucleotide reductase) and the strong inhibition of ribonucleotide reductase by cladribine (a weak inhibitor of DNA polymerases) into one molecule that is a strong inhibitor of both DNA elongation and ribonucleotide reductase. The relative importance of clofarabine’s actions to the antitumor activity of clofarabine is not known, but it is likely that both targets significantly contribute to its cell-killing action.
16.5.2 Repair of DNA Containing Clofarabine
The removal of clofarabine from the 3′-end of DNA chains by 3′-exonucleases that are associated with the DNA replication complex is an important part of the mechanism of action of this compound that has not yet been evaluated. If clofarabine is quickly removed from the 3′-terminus of the DNA chain, DNA synthesis can continue normally, although the polymerase would still have a chance to incorporate another molecule of clofarabine in the repair site, resulting in a futile incorporation and removal cycle. The incorporation of two or more clofarabine residues sequentially in the DNA may be harder to repair than single incorporations and could represent a greater block to DNA synthesis. Once the DNA polymerase extends beyond the incorporated analog, DNA synthesis can continue normally. It is not known whether clofarabine that is successfully incorporated into the DNA chains (beyond the replication sites) results in DNA that cannot function normally. However, Hentosh and colleagues [53, 54] demonstrated that the random incorporation of cladribine into DNA can disrupt transcriptional regulation by altering DNA binding of TATA-binding protein and can have deleterious effects on cell function. It is likely that the random incorporation of clofarabine would have similar effects. However, if there are negative consequences to the incorporation of clofarabine into DNA chains, these actions are secondary in importance to the cell-killing mechanism of action of clofarabine.
16.5.3 Induction of Apoptosis
The inhibition of replication in cells normally leads to the turning on of replication checkpoint pathways. Stalled replication forks can threaten DNA replication fidelity, and cells respond to replication blocks by triggering checkpoint pathways that monitor replication fork progression [55]. This monitoring of DNA synthesis operates normally during low-intensity replication stress and is required for tumor cells to resume cell cycle progression. However, during chronic or high-intensity replication stress, stalled forks do not restart after removal of the stressor, resulting in irreversible S-phase arrest, possible mitotic catastrophe, and cell death.
The inhibition of DNA synthesis and the DNA damage caused by clofarabine resulted in the induction of apoptosis in CEM and HCT116 cells [56–58]. Replication stress induced by the treatment of CEM cells with 10 μM clofarabine leads to Chk1 phosphorylation, which is accompanied by Chk1 downregulation, concomitant apoptosis, and cell death [5]. Incubation with clofarabine resulted in a dose- and time-dependent downregulation of Bcl-XL, Mcl-1, and Cdc25A proteins, but it did not affect Cdc25C [57]. Treatment with clofarabine also resulted in the dephosphorylation of Akt and its downstream effectors (Bad and FKHRL1). In addition, there was a marked increase in the population of cells in G1/S and early S phase. Clofarabine led to more apoptotic cell death than did fludarabine, suggesting that DNA damage caused by clofarabine is more easily recognized by the surveillance systems that initiate apoptotic cascades [51].
16.5.4 Activity Against Non-proliferating Cancer Cells
Clofarabine is also active against quiescent human lymphocytes and monocytes [52]. Treatment of these cells with clofarabine induced DNA strand breaks, which were markedly reduced by supplementation of the culture medium with dCyd. Clofarabine also interfered with mitochondrial integrity in primary CLL cells [59], which led to the release of the proapoptotic mitochondrial proteins cytochrome C and apoptosis protein-activating factor (Apaf-1). The activity of clofarabine was similar to that of cladribine, and both agents induced mitochondrial damage more effectively than did fludarabine. The investigators proposed three mechanisms that may be responsible for the mitochondrial damage in non-proliferating CLL cells: (1) incorporation into nuclear DNA, resulting in DNA damage and inducing an apoptotic cascade; (2) binding to Apaf-1, resulting in the induction of apoptosis [60]; and (3) direct interference with mitochondrial function. They felt that mitochondrial damage caused by clofarabine or cladribine resulted from all three effects, whereas mitochondrial damage caused by fludarabine resulted from only nuclear DNA damage and Apaf-1 binding. They suggested that the direct activity against mitochondrial function could be due to the binding of clofarabine and cladribine nucleotides to proteins that are known to regulate mitochondrial functions. Clofarabine triphosphate was able to activate Apaf-1-dependent caspase activity in CLL cell extracts with kinetic parameters that were identical to those of dATP [60].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


