Fig. 27.1
Comparison of overall survival and surrogate endpoints
Disease-Free Survival (DFS)
Disease-free survival (DFS) is defined as the time from randomization until recurrence of tumor or death from any cause. DFS is a common surrogate endpoint used in breast cancer clinical trials, especially in the adjuvant setting following surgery or radiotherapy. In a situation where the survival is prolonged, it may not be practically feasible to measure the overall survival and the DFS may be a preferred endpoint in this scenario and can be an early indicator of improved survival. Patients without recurrent disease may be detected by DFS. Many of the hormonal therapies and cytotoxic chemotherapies that have been approved by the regulatory agencies have employed DFS as the primary endpoint in the exploratory and confirmatory clinical trials. It is essential that while designing a clinical trial with DFS as endpoint, the DFS definition should be clearly stated in the trial protocol. The schedule for follow-up must be stated without any ambiguity to avoid unscheduled visits, which can introduce bias into the study. Bias can be introduced into the study if the number of follow-up visits is greater in one arm of the study due to the development of toxicity in the other arm.
Although deaths that occur in breast cancer clinical trial can be due to disease recurrence, there is always a possibility of deaths being noted without any prior documentation of tumor progression. It is common practice to consider all deaths as recurrences to reduce the amount of bias. However, there is a possibility of overestimation of DFS if patients survive for a long period of time. DFS identifies the proportion of patients without disease recurrence [11, 13].
One of the major challenges in using DFS as the primary endpoint is the variable definition of DFS in different breast cancer trials. Some of the events that are included under breast cancer DFS endpoints by most trialists are contralateral breast cancer that includes invasive lobular or ductal carcinoma, such as ductal carcinoma in situ and in situ carcinoma and deaths from other causes. When there is variability in breast cancer DFS definitions, it becomes less appropriate to compare the different trial results and make meaningful conclusions. There is also a possibility of a treatment being deemed as improved by one particular definition of DFS but not by another [13].
A panel of experts comprised of medical oncologists, biostatisticians, and other scientific experts from several reputed institutions in the USA and Canada convened to standardize the guidelines for endpoint definitions in breast cancer trials. The panel suggested the replacement of DFS with a more specific endpoint termed invasive disease-free survival (IDFS) for early breast cancer adjuvant trials [13]. Listed below are the events included under IDFS:
Ipsilateral invasive breast tumor recurrence (IIBTR): invasive breast cancer involving the same breast parenchyma as the original primary
Regional invasive breast cancer recurrence: invasive breast cancer in the axilla, regional lymph nodes, chest wall, and skin of the ipsilateral breast
Distant recurrence: metastatic disease breast cancer that has either been biopsy confirmed or clinically diagnosed as recurrent invasive breast cancer
Death attributable to any cause, including breast cancer, non-breast cancer, or unknown cause
Contralateral invasive breast cancer
Second primary non-breast invasive cancer
Distant metastasis is the most critical parameter that influences the survival of the patient. Hudis et al. have proposed a novel endpoint termed distant disease-free survival, under which ipsilateral breast tumor recurrence, regional invasive recurrences, contralateral breast cancer, and all in situ carcinomas are avoided as events, since they have minimal potential to affect survival [13]. There is a strong correlation between distant disease recurrence and death. Distant disease-free survival includes only distant recurrence and death due to breast cancer and non-breast cancer deaths.
Objective Response Rate (ORR)
Objective response rate (ORR) is defined as the proportion of patients with tumor size reduction of a predefined amount and for a minimum time period [12]. The ORR is the proportion of patients with a best overall response of confirmed complete (CR) or partial (PR) response. ORR is considered a direct measure of antitumor drug activity, but not a direct measure of clinical benefit [14]. It is important to state the definition of response without any ambiguity in the clinical trial protocol. It is not pertinent to include stable disease under objective response rate, as stable disease can reflect the natural history of the disease, thereby obfuscating the trial results. Since ORR can be achieved even in a short span of time in certain cases, it is one of the endpoints that is selected for accelerated approval. ORR was formerly considered an adequate endpoint for assessment and approval of an anticancer agent by the FDA. However, the subsequent realization that endpoints such as OS are more reflective of the drug’s benefit made the regulatory authorities seek for OS in clinical trial protocols instead of ORR. Nevertheless, ORR is still considered a major endpoint in phase II clinical trials of breast cancer therapies [2].
Response rate can be most accurately determined only with the help of imaging technology such as X-ray, CT scan, or MRI using RECIST (Response Evaluation Criteria in Solid Tumors) criteria. The RECIST criteria are a standard set of guidelines that have been designed by the EORTC (European Organization for Research and Treatment of Cancer) and NCI (National Cancer Institute) of the USA and Canada to assess the progression of tumors [15]. The RECIST criteria were originally proposed in 2000 and were modified in 2009 to become known as RECIST 1.1 [16]. The FDA generally defines ORR as the sum of partial responses plus complete responses (CRs). According to RECIST 1.1 criteria, in a target lesion, CR is defined as the disappearance of all target lesions, while partial response refers to at least a 30 % decrease in target lesions. Progressive disease is a 20 % increase in the sum of target lesions, while stable disease is lack of sufficient shrinkage to qualify for partial response or progressive disease [16]. Since the treatment effect is directly attributable to drug activity, single-arm trial design may be applied when using ORR as an endpoint.
The RECIST criteria are useful in all situations when assessment of anatomical tumor burden is to be made and its response to therapy. The clinical significance of ORR must be determined by performing a risk–benefit analysis to ascertain the magnitude and duration of the effect [12]. Bruzzi et al. analyzed randomized trials which compared a standard FEC (fluorouracil, epirubicin, and cyclophosphamide) regimen with a dose-intensified FEC regimen and showed that tumor response is a highly significant predictor of survival [17]. However, in another analysis by Burzykowski et al., no endpoint could be identified as a good surrogate for overall survival [18].
Progression-Free Survival (PFS)
Progression-free survival (PFS) is defined as the time from randomization or treatment initiation until tumor progression or death. Since it usually requires a shorter follow-up period and smaller sample size than studies measuring overall survival (OS), and is not confounded by subsequent therapies, it is often used as a surrogate marker for accelerated approval of drug therapies. In situations where the deaths due to cancer are higher, as in advanced breast cancer, progression-free survival is a better indicator than time to tumor progression.
A study by Burzykowski et al. showed that tumor response is predictive of PFS in patients with advanced breast cancer and thus can be used as a surrogate marker of PFS. However, the authors rightly conclude that analyses regarding the validation of surrogate endpoints are “specific to well-defined disease, clinical outcome, and treatment” [18]. PFS is currently considered the most sensitive parameter for evaluation of the efficacy of a drug [19]. It is not surprising that a literature search across the clinical trial registry data of breast cancer phase III trials showed that PFS was the most common primary endpoint chosen, as illustrated in Fig. 27.2.
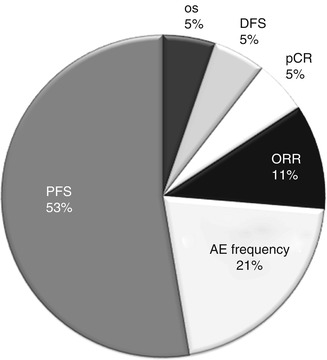
Fig. 27.2
Primary endpoints used in phase 3 clinical trials in breast cancer. Data is based on the list of studies registered in the clinicaltrials.gov registry in the year 2012 that are active. PFS progression-free survival, OS overall survival, DFS disease-free survival, pCR pathologic complete response, ORR objective response rate, AE adverse event
The advantage of PFS includes objective and quantitative assessment of the disease outcome. Moreover, PFS is not affected by crossover treatment. The limitations of PFS include frequent assessment of disease progression at reasonable intervals through various investigations including radiological evaluation in both the study arms. This may add to the cost of the trial. Moreover, the definition of PFS may differ across trials, as there are no standard regulatory criteria to define progression of disease. Hence it is not considered a statistically validated surrogate endpoint for survival. Further, missing data in the trial may obscure the analysis of PFS. This could be overcome by assigning follow-up visits in the earlier stage to assess progression, while censored visits may be assigned after radiological examination reveals no progression in the disease. In addition, assessment bias is a major disadvantage of PFS as progression cannot be precisely measured. This could be overcome by randomized and blinded study design [6].
In the present scenario, PFS is gaining preference over overall survival (OS) as the primary endpoint in randomized clinical trials conducted in solid tumors. This could be attributed to the availability of earlier outcome with PFS compared to OS. Moreover, PFS is not affected by second-line treatments for cancer [20]. PFS is being validated as a surrogate endpoint in various cancers including breast cancer and colorectal cancer. In advanced colorectal cancer, PFS has been found to be a valid surrogate as opposed to OS following the completion of first-line chemotherapy. In a literature search done on metastatic colorectal cancer after first-line chemotherapy, appraising PFS as a potential surrogate endpoint compared to OS, a promising correlation was shown with OS [21]. In contrast, PFS has not been validated as a surrogate endpoint in advanced breast cancer so far. Recent investigations have shown that tumor response is an acceptable surrogate for PFS in patients with advanced breast cancer since the correlation seems to be reasonably good [18]. However, the success of using PFS as a potential endpoint to interpret clinical outcome needs to be explored [9].
Time to Tumor Progression (TTP)
Apart from progression-free survival, time to tumor progression is one of the most common endpoints used in the field of breast cancer clinical trials. Time to tumor progression (TTP) is defined as the time from randomization to time of progressive disease and censors deaths that occur before progression of disease. Since PFS includes deaths, it is preferable to TTP as an endpoint in breast cancer clinical trials. While PFS assumes that patient deaths are related to tumor progression, TTP discounts deaths altogether. TTP is especially appropriate in a situation where the majority of deaths are not related to tumor progression [6].
It has been found that since 1975 there has been an increase in the number of clinical trials conducted with the use of PFS/TTP as primary endpoints, especially in breast, colorectal, and non-small cell lung cancers [22]. For drug approval, the FDA considers both PFS and TTP as the primary endpoints for evaluating efficacy. However, the choice among the two is based on the magnitude of the effect and the risk–benefit profile of the drug product. During analysis of TTP deaths without documented progression are censored, which may lead to biased estimates. This can be overcome with PFS, which includes death in analysis and takes into consideration treatment effects not mediated through tumor burden. Hence PFS is preferred over TTP as a regulatory endpoint [23]. However, while assessing TTP or PFS, patients should be evaluated at regular intervals in all treatment arms including assessment of all disease sites. To reduce bias further, similar assessment techniques should be carried out during each follow-up visit. Nevertheless, a statistically significant difference in TTP or PFS between treatment arms need not inevitably mean clinical benefit [12]. Bowater et al. showed that “the time period between the start of treatment and disease progression (i.e., time to progression) has a strong tendency to extend, by roughly the same amount, the period between the start of treatment and death (i.e., overall survival)” [24].
Time to Treatment Failure (TTF)
Time to treatment failure (TTF) is defined as the time from randomization to discontinuation of treatment for any reason, including disease progression, treatment toxicity, and death. Sometimes TTF is mistakenly defined as the time from study entry to progression of disease or death. However, TTF being a composite endpoint comprises subjective assessment of symptoms in addition to reasons for discontinuation of therapy [25]. A prospective multicenter randomized study done to compare the safety and effectiveness of vinorelbine and melphalan in patients with anthracycline-refractory advanced breast cancer included TTF as one of the efficacy endpoints along with time to disease progression, survival, tumor response rates, and quality of life [26]. To be considered as a regulatory endpoint, the parameter assessed should differentiate between drug efficacy and adverse effect, which is lacking in TTF. Hence the FDA does not recommend TTF as a regulatory endpoint, and it is not considered for drug approval [6].
Pathologic Complete Response (pCR)
There has not been a uniform consensus as of yet on the definition of pathologic complete response (pCR). Some investigators define pCR as the absence of residual cancer in the breast and regional lymph nodes at the time of definitive surgery, whereas others have defined pCR as a complete response in the breast, irrespective of axillary nodal involvement [27–30]. As per the FDA draft guidance document, pCR is defined as the absence of any residual invasive cancer on hematoxylin and eosin evaluation of the resected breast specimen and all sampled ipsilateral lymph nodes following completion of neoadjuvant systemic therapy. pCR is especially useful in the setting of evaluating neoadjuvant systemic chemotherapy, and several trials have utilized pCR as an endpoint in these settings [31]. Since the pathologists are playing a crucial role in evaluating pCR, it is required that they be blinded to the treatment arms to avoid bias.
The Collaborative Trials in Neoadjuvant Breast Cancer (CTNeoBC) carried out a large-scale meta-analysis involving more than 13,000 patients to assess the correlation between pCR and DFS/OS and to also determine the types of breast cancer that pCR is most likely to predict clinical benefit [32]. The study showed that individual patients who attain a pCR, defined as either ypT0ypN0 or ypT0/isypN0, have a more favorable long-term outcome and the data show comparable OS. Thus pCR has an association with long-term outcomes such as OS. In the coming years as our understanding of pCR as an endpoint improves, it is more likely to be used as a primary endpoint in regulatory clinical trials.
Clinical Benefit Rate (CBR)
Clinical benefit rate (CBR) is the proportion of patients with a best overall response—of complete response (CR), partial response (PR), or stable disease (SD)—lasting more than 24 weeks as defined in RECIST 1.1. Patients are followed up for the duration of the study and for an expected average of every 8 weeks after randomization. Disease control rate (DCR) and CBR are defined as the percentage of patients with advanced or metastatic cancer who have achieved complete response, partial response, and stable disease to a therapeutic intervention in clinical trials of anticancer agents [33].
Patient-Reported Outcomes for Global Health Status/QOL
Patient-reported outcomes, such as time to definitive deterioration in global health status/quality of life (QOL), offer additional valuable information about the nature of the study drug that complements the other conventional endpoints [34]. The major disadvantage with these endpoints is the absolute requirement for randomization and blinding to avoid bias in assessment of outcomes. One should be careful to distinguish the symptoms that arise due to the malignancy and those that arise due to drug toxicity [12]. Patient-reported outcomes such as QOL gain importance in the context of therapies which are not expected to offer any substantial benefit on patient survival. Although not considered important enough to warrant itself as a primary endpoint, QOL is not infrequently measured as one among the several secondary endpoints measured [1]. There is a remarkable heterogeneity in the types of QOL scales used by different groups. Some of the scales used to assess QOL in clinical trials with breast cancer include the European Organization for Research and Treatment of Cancer QOL questionnaires (EORTC QLQ-C30, EORTC QLQ-BR23), the Functional Assessment of Cancer Therapy questionnaires (FACT-G, FACT-B) and its subscales, the DBCG-89 questionnaire, the Medical Outcomes Study 36-Item Short-Form Health Survey (MOS-SF-36), the Hospital Anxiety and Depression Scale (HADS), and the International Breast Cancer Study Group (IBCSG) approach [35]. Due to the shortfall in the methodologic standards applied in evaluating QOL in clinical trials of breast cancer, assessment of QOL appears to have limited benefit in choosing the right therapy for the patient [36]. There is a definite need for more consistency among the different groups so as to make the results of different studies comparable with each other [37].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


