Langerhans cell histiocytosis (LCH) is a disease caused by clonal proliferation of CD1a+/CD207+ cells that is characterized by a spectrum of varying degrees of organ involvement and dysfunction. Treatment of LCH is risk adapted; patients with single lesions may respond well to local treatment, whereas patients with multi-system disease and risk-organ involvement require more intensive therapy. Although survival for patients without organ dysfunction is excellent, mortality rates for patients with organ dysfunction may reach 30% to 40%. For patients with low-risk disease, although cure is almost universal, disease reactivation rates are in excess of 30%.
Key points
- •
Langerhans cell histiocytosis (LCH) is a neoplasm of myeloid origin characterized by a clonal proliferation of CD1a+/CD207+ cells. In a little more than half of the cases, BRAF mutations, predominantly encoding BRAF V600E, are identified; mutations of other members of the MAPK/ERK pathway, such as MAP2K1 or ARAF , are present in another 10% to 25%.
- •
LCH affects individuals of all ages, although infants more often present with multisystem disease. The disease can affect many tissues and organs of the head and neck. Bony lesions are most common; but the skin, lymph nodes, and brain can also be involved.
- •
Patients with involvement of only one organ system can often be treated with surgery alone and have excellent outcomes. Patients with multisystem disease, especially with risk-organ involvement, need multimodality treatment and have variable prognoses.
- •
Treatment with BRAF inhibitors has shown to induce complete and durable responses, and the role of BRAF and MEK inhibitors is currently being investigated.
- •
LCH neurodegeneration, a devastating long-term complication of LCH, represents one of the major challenges in clinical and translational research of LCH.
Introduction
Langerhans cell histiocytosis (LCH) is a disease characterized by clonal proliferation of CD1a+/CD207+ myeloid dendritic cells that presents at all ages and with different degrees of systemic involvement. Almost any organ can be affected, and the clinical presentation reflects the tissue-specific inflammatory phenomena. For several decades, LCH has been considered to be a reactive clonal proliferation of Langerhans cells; however, in recent years, LCH has been defined as a neoplasm of myeloid origin, with a significant inflammatory component that defines some of the acute and long-term manifestations. This evolution from an immune disorder to a neoplasia has reframed the disease and opened the door for the development of targeted therapies.
Introduction
Langerhans cell histiocytosis (LCH) is a disease characterized by clonal proliferation of CD1a+/CD207+ myeloid dendritic cells that presents at all ages and with different degrees of systemic involvement. Almost any organ can be affected, and the clinical presentation reflects the tissue-specific inflammatory phenomena. For several decades, LCH has been considered to be a reactive clonal proliferation of Langerhans cells; however, in recent years, LCH has been defined as a neoplasm of myeloid origin, with a significant inflammatory component that defines some of the acute and long-term manifestations. This evolution from an immune disorder to a neoplasia has reframed the disease and opened the door for the development of targeted therapies.
Biology
The pathogenic cells are known to originate from a myeloid-derived precursor and are uniformly characterized by activation of the MAPK/ERK signaling pathway; ERK activation is documented in all cases. In up to two-thirds of cases, pathway activation is secondary to a somatic mutation in BRAF (BRAF V600E ) ; in other cases, mutations in MAP2K1 or less frequently in other members of the pathway, such as ARAF , have been described. About one-quarter of cases have no known genomic abnormalities.
Epidemiology
The estimated incidence of LCH is 8.9 cases per million children younger than 15 years per year, with a median age at diagnosis of 3 years old. The causes and risk factors for developing LCH are unclear. However, the unique patterns of presentation, ranging from localized bone lesions with spontaneous regression to disseminated forms with multiorgan involvement, suggest a complex pathogenesis. Familial associations, particularly the observation of increased incidence in monozygotic twins of affected patients, have suggested the presence of a germline predisposition at least for a proportion of cases. Also, population-based studies have shown differences in the incidence of disseminated LCH by race and ethnic group; a higher incidence has been reported for Hispanics and a lower incidence for blacks. Studies have also shown a correlation with maternal and neonatal infections, lack of childhood vaccinations, family history of thyroid disease, in vitro fertilization, and feeding problems and transfusions during infancy. Finally, lower socioeconomic conditions have been associated with an increased incidence of disseminated LCH.
Pathology
Since pathologic Langerhans cells activate other immunologic cells, microscopic examination of diseased tissue shows eosinophils, neutrophils, lymphocytes, and histiocytes in addition to the LCH cells; this appearance is what has been traditionally described as eosinophilic granuloma. Abscesses and necrosis may be present. LCH cells are large, oval , and mononuclear, with a prominent nucleus and eosinophilic cytoplasm. They do not have dendritic cell processes like cutaneous Langerhans cells. They stain positive for protein S-100, CD1a, and CD207 (langerin) and contain cytoplasmic rod-shaped inclusions called Birbeck granules. A diagnosis of LCH is made by typical positive staining with CD1a or CD207.
With the development of new technology for accurate detection of cell-free DNA, BRAF V600E mutation analysis in plasma and urine has shown to be an effective tool for diagnosis and monitoring of disease activity in patients with LCH.
Clinical presentation
Classically, LCH was defined as 3 distinct diseases; eosinophilic granuloma, Hand-Schüller-Christian disease, and Abt-Letterer-Siwe disease were different clinical descriptions within the same spectrum of progressive system involvement. Eosinophilic granuloma , whether solitary or multifocal, is found predominantly in older children as well as in young adults, with a peak incidence between 5 and 10 years of age. Eosinophilic granuloma is the most common form of LCH, accounting for approximately 60% to 80% of the diagnoses. Hand-Schüller-Christian disease was historically described as a clinical triad of lytic bone lesions, exophthalmos caused by orbital involvement, and diabetes insipidus (DI). It is most commonly described in the first 4 to 7 years of life, and it may account for approximately 15% to 40% of all LCH cases. Abt-Letterer-Siwe disease is the most severe manifestation of LCH, albeit rare. Typically, patients are less than 2 years of age and present with a scaly seborrheic rash, ear discharge, and signs of severe systemic involvement with symptoms such as cytopenias, pulmonary dysfunction, lymphadenopathy, or hepatosplenomegaly. Nowadays, this old terminology has been replaced by a classification system that is based on the site of lesions, number of involved sites (single or multisystem/local or multifocal), and whether the disease involves risk organs (hematopoietic system, liver, or spleen). This classification is the basis for the risk-adapted treatment used in the LCH-III protocol discussed later ( Table 1 ).
| Clinical Group | Involved System | Involved Organs |
|---|---|---|
| 1 | Multisystem risk patients | Any risk a organ involvement |
| 2 | Multisystem low-risk patients | ≥2 Organ without risk a organ involvement |
| 3 |
| ≥2 Lesions in one organ or in special site b |
| — | Single system Unifocal or localized | 1 Lesion in one organ |
a Risk organs consist of lung, liver, spleen, bone marrow, or hematological dysfunction.
b Special sites are intracranial soft tissue extension or vertebral lesions with intraspinal soft tissue extension.
Sites of involvement
All organs can be affected by LCH; therefore, a comprehensive evaluation is indicated. The most commonly involved organs and systems are highlighted next:
Bone
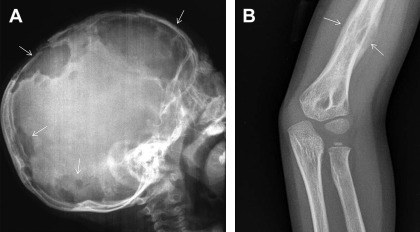
Bone is the most commonly affected system; bone lesions are present in approximately 80% of patients with LCH. The most common site of involvement is the skull (27%), followed by the femur (13%), mandible (11%), and pelvis (10%). Mostly, radiographic studies typically show lytic lesions, especially punched-out lesions in the skull without marginal sclerosis or periosteal reaction ( Fig. 1 ). Pain and tumor formation in a localized area of bone is a very common presentation of LCH. In the skull, the lesions are usually soft and tender to touch. Skull lesions may include a soft tissue mass pressing on the dura, but severe intracranial extension is rare. Involvement of the skull base is also very common in LCH; typical locations include the bones of the orbit or the temporal bone (typically the mastoid). In these cases, otitis media or externa are common presenting signs. Involvement of the vertebral bodies is also common, and the presence of a vertebra plana is frequent.
Skin
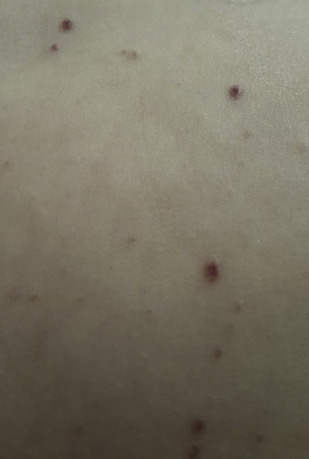
The most common skin lesions are seborrheic eczema, which typically occurs in infants, and a neonatal form characterized by disseminated brown to red papules with common central ulceration ( Fig. 2 ). Isolated skin involvement usually carries a good prognosis, with approximately 60% chance of regression. However, close monitoring is required, as reactivation or progression to multisystem involvement has been observed in up to 40% of cases.
Neuroendocrine and Central Nervous Systems
DI caused by involvement of the pituitary stalk occurs in approximately 25% of cases, usually after therapy. LCH is also a common diagnosis in patients with DI of unknown cause, and almost all patients with DI caused by LCH have involvement of other organs concurrently or subsequently. Anterior pituitary involvement, commonly represented by growth hormone deficiency, is also a common complication and highlights the importance of a comprehensive endocrine monitoring in patients with LCH. Mass lesions of the gray or white matter are less frequent (1%). Involvement of the sphenoid, orbital, ethmoid, zygomatic, or temporal bones confers a higher (25% overall) risk for central nervous system (CNS) involvement (CNS-risk lesions), including late neurodegeneration (see later discussion), an inflammatory phenomenon of unclear pathogenesis that is characterized by cerebellar dysfunction and neurocognitive deficits.
Pulmonary
Involvement of the lungs usually occurs in the context of multisystem involvement, commonly limited to young children. Patients usually present with pulmonary dysfunction including tachypnea, dyspnea, and cough. Radiographic findings are typical for the presence of a reticulonodular pattern with bullae formation. Isolated pulmonary involvement is a rare presentation that is almost exclusively found in adults with a smoking habit (discussed later).
Hematopoietic System
The presence of hematopoietic dysfunction in the form of cytopenias is a poor prognostic sign. It occurs in the context of multisystem involvement, more frequently in young children. Its pathophysiology is multifactorial, including direct involvement of the bone marrow as well as peripheral destruction caused by hypersplenism from Langerhans cell infiltrates in the spleen.
Hepatobiliary System
Liver involvement, which typically occurs in infants with multisystem disease, also carries a poor prognosis. Patients present with hypoalbuminemia, edema, hepatomegaly, or conjugated hyperbilirubinemia. A well-described complication of hepatic involvement is the development of sclerosing cholangitis and hepatic fibrosis, which can result in liver failure and need for liver transplantation.
Treatment of Langerhans cell histiocytosis
The treatment of LCH over the years has reflected the changing concepts of the disease process. Indeed, the difficulties in developing more effective therapies are linked to the deficiencies in the understanding of the pathogenesis of LCH. Retrospective studies of Lahey and Komp and colleagues showed that, although many organs can harbor proliferating pathogenic cells, only if organ function was disrupted was such involvement of prognostic significance. Patients could then be stratified into different risk categories based on the extent of their disease and the degree of organ dysfunction. Patients with single-system disease confined to a single site usually require only local therapy or observation. Patients with more extensive disease require systemic therapy; several groups have explored risk-based approaches, which are summarized next.
Histiocyte Society’s Langerhans Cell Histiocytosis Studies
Langerhans Cell Histiocytosis-I (LCH-I)
The first international randomized trial for multisystem LCH (MS-LCH), LCH-I (1991–1995), randomized patients to receive weekly vinblastine or etoposide every 3 weeks for 24 weeks. Overall survival was close to 80%, with no significant differences by treatment arm. Involvement of the hematopoietic system, lung, liver, and spleen, age at diagnosis less than 2 years old, and poor response at 6 weeks were significantly associated with an adverse outcome. In comparison with the contemporaneous DAL-HX 83 study, which explored a more intensive and longer regimen, LCH-I showed a lower 6-week response rate (50% vs 80%) and a higher reactivation rate (50% vs 30%).
Langerhans Cell Histiocytosis-II (LCH-II)
The LCH-II study (1996–2001) followed on the LCH-I findings and explored early intensification through a randomized design that investigated the addition of etoposide (arm B) to a standard 6-week induction with prednisone and vinblastine and continuation with 6-mercaptopurine and every-3-week pulses of prednisone and vinblastine for a total of 24 weeks of therapy (arm A). Both arms produced similar outcomes in terms of 6-week response rates (63% arm A vs 71% arm B), 5-year survival probability (74% vs 79%), and disease reactivation rates (46% both arms); however, the more intensive arm B resulted in reduced mortality for patients with risk organ involvement. The LCH-II study also showed that patients younger than 2 years without risk-organ involvement have excellent response rates and a 100% survival, and patients with risk-organ involvement and poor response at 6 weeks have the highest mortality. Those findings helped refine the risk-stratification and treatment for its successor study, the LCH-III.
Langerhans Cell Histiocytosis-III (LCH-III)
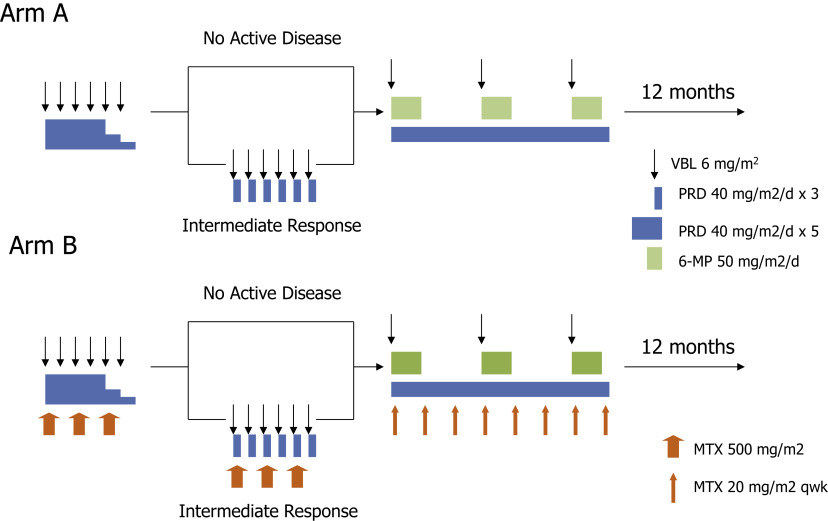
The LCH-III study (2001–2008) was built following a risk-adapted model with the main objectives of investigating the impact of the addition of methotrexate on the outcome of patients with risk-organ involvement and the effect of therapy prolongation on decreasing the incidence of disease reactivation for patients with multisystem disease. The 3 risk groups are depicted in Table 1 . Patients with MS-LCH with risk-organ involvement (group 1) received standard induction with prednisone and vinblastine and continuation with the same drugs with the addition of 6-mercaptopurine for a total duration of 12 months and were randomized to the addition of methotrexate; patients with active disease at 6 weeks received a modified reinduction ( Fig. 3 ). The outcome was similar in both arms, with a 5-year survival probability of 84%, response rates of 71%, and reactivation rates of 27%. Historical comparisons revealed superior outcomes compared with LCH-I and LCH-II in terms of survival and reactivation rates. The 3-year cumulative incidence of DI was also similar in both arms (8% in standard arm and 9% in methotrexate arm).
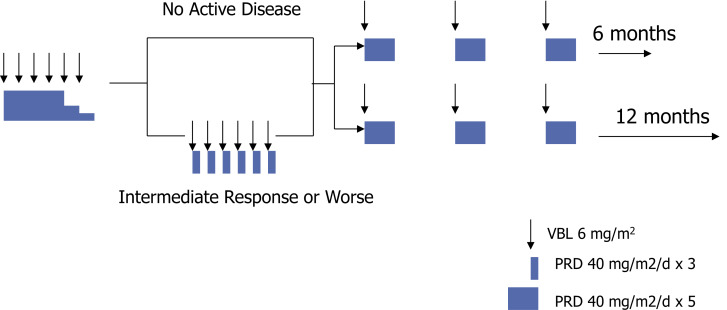
The outcome for patients with MS-LCH without risk-organ involvement is excellent, with survival rates close to 100% across studies. However, in the LCH-I and LCH-II studies, almost half of these patients sustained a disease reactivation. Based on the lower reactivation rates noted in the DAL-HX-83 study, which prolonged treatment for 12 months, the LCH-III protocol investigated the effect of treatment duration for this group of patients (group 2) and randomized them to a standard regimen of vinblastine and prednisone for 6 or 12 months ( Fig. 4 ). Longer treatment resulted in a significantly lower 5-year reactivation rate (37% vs 54%, P = .03). The 3-year cumulative incidence of DI was 12% in both arms.
Group 3 included patients with single-system multifocal disease (mostly multifocal bone) and patients with single bone lesions in special sites. Special sites were defined as involvement of the craniofacial bones with intracranial extension and patients with vertebral lesions with intraspinal soft tissue extension. Those patients were treated with a standard 6-month regimen of prednisone and vinblastine.
Langerhans Cell Histiocytosis-IV
The LCH-IV protocol represents a major international effort to integrate the most relevant clinical questions in a prospective trial, of which the overarching objectives are as follows: (1) to improve survival for patients with risk-organ involvement by early switching to intensive nucleoside-analogue–based therapy; (2) to investigate whether further prolongation of therapy will decrease reactivation rates; and (3) to investigate the incidence, pathogenesis, and treatment of LCH-induced neurodegeneration. Thus, the LCH-IV study provides an excellent framework on which major therapeutic questions can be addressed. Because of the extreme complexity of the disease presentations and course scenarios, the LCH-IV study consists of 7 strata:
Stratum I
This stratum includes the first-line treatment of patients with MS LCH (group 1) and patients with single-system (SS) LCH with multifocal bone or CNS-risk lesions (group 2). Group 1 patients will undergo a double randomization; all will receive a standard prednisone/vinblastine regimen and will be randomized to 12 versus 24 months and to the addition of 6-mercaptopurine. Group 2 patients will be randomized to receive the standard prednisone/vinblastine regimen for 6 versus 12 months.
Stratum II
This stratum includes the second-line treatment of nonrisk patients (patients without risk-organ involvement who fail first-line therapy or have a reactivation after completion of first-line therapy). Patients will be treated with a combination of vincristine and low-dose cytarabine (ara-C) for 24 weeks, after which they will be randomized to indomethacin or a combination of oral 6-mercaptopurine and methotrexate as maintenance.
Stratum III
This stratum includes salvage treatment of risk LCH (patients with dysfunction of risk organs who fail first-line therapy). This group of patients with high-risk disease has the worse outcome; in order to improve survival, an early switch (in first 6 weeks) to this salvage arm will be indicated. Treatment includes an intense regimen based on high-dose cytarabine and cladribine.
Stratum IV
This stratum includes stem cell transplantation for risk LCH (patients with dysfunction of risk organs who fail first-line therapy).
Stratum V
This stratum includes monitoring and treatment of isolated tumorous and neurodegenerative CNS-LCH. This stratum will study the efficacy of cladribine in isolated tumorous CNS-LCH and intravenous immunoglobulin and cytarabine in neurodegenerative CNS-LCH treatment.
Stratum VI
This stratum includes the natural history and management of other SS-LCH (patients who do not need systemic therapy at the time of diagnosis).
Stratum VII
This stratum includes the long-term follow-up. (All patients irrespective of previous therapy will be followed for reactivation or permanent consequences once complete disease resolution has been achieved and the respective protocol treatment completed.)
The definition of risk organ involvement is depicted in Box 1 ; importantly, in LCH-IV, lung involvement is no longer considered an adverse prognostic factor as it was in earlier studies.
Hematopoietic involvement : (with or without bone marrow involvement a )
At least 2 of the following:
- 1.
Anemia: hemoglobin less than 100 g/L (<10 g/dL), infants less than 90 g/L (<9.0 g/dL), not from other causes (eg, iron deficiency)
- 2.
Leukocytopenia: leukocytes less than 4.0 × 10 9 /L (4000/μL)
- 3.
Thrombocytopenia: platelets less than 100 × 10 9 /L (100.000/μL)
- 1.
Spleen involvement: enlargement greater than 2 cm below costal margin in the midclavicular line b
Liver involvement: one or more of the following
- 1.
Enlargement greater than 3 cm below costal margin in the midclavicular line b
- 2.
Dysfunction (ie, hypoproteinemia <55 g/L, hypoalbuminemia <25 g/L, not from other causes)
- 3.
Histopathologic findings of active disease
- 1.
a Bone marrow involvement is defined as presence of CD1a-positive cells on marrow slides.
b Enlargement in centimeters below the costal margin as assessed by physical examination.
Related posts:
 Dedication
Dedication
 Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
 Pathogenesis of Hemophagocytic Lymphohistiocytosis
Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


