Fig. 16.1
ALL: PAS reaction. Note the block-like reaction product
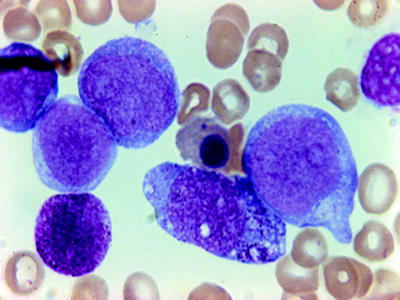
Fig. 16.2
AML (FABM0). Blasts with no differentiation. Peroxidase negative but with positive myeloid antigens by flow cytometry
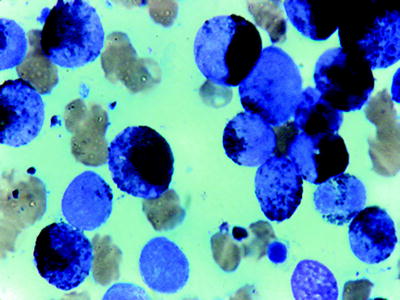
Fig. 16.3
AML (myeloperoxidase stain). Black reaction product with Auer rods
There are three instances in which the diagnosis of acute leukemia can be made on the basis of histologic material. The first occurs when there is an abundance of reticulin in the bone marrow, resulting in a so-called dry-tap [3]. This can be present in any of acute leukemias and is recognized most commonly associated with acute megakaryoblastic leukemia. Although touch preparation smears may be of help in such cases, well-prepared hematoxylin and eosin (H&E) stains of paraffin-embedded material, PAS, chloroacetate esterase stains, and the immunologic methods identifying antigens on cell surface (CD41, CD61 or Factor VIII) can often establish the diagnosis. The second example is hypocellullar (hypoplastic) acute myeloid leukemia in which bone marrow aspirations from several sites and biopsy from one site are necessary for diagnosis [4]. The correct diagnosis depends on a representative biopsy specimen. The third example is that of an uncommon localized extramedullary mass of cells of the granulocytic–monocytic series (myeloid sarcomas) [5]. These tumors can be seen in an established diagnosis of AML, either at presentation or as the first manifestation of relapse.
Diagnosis of Acute Leukemias
The most widely used morphologic classification of acute leukemia for over two decades was that proposed by the French–American–British (FAB) Cooperative group [6]. The classification is based exclusively on the morphology of the bone marrow and peripheral blood leukemic cells in the Ramonowsky-stained smears. The initial intent was to separate ALL and AML into easily identifiable cell types. In attempting to define boundaries between overlapping groups, major attention is given to the predominant cell type present. An essential cytochemical stain is one that demonstrates MPO, an enzyme restricted to the primary granules of granulocytes and monocytes [7]. FAB classification has stood the test of time, since only a few cases do not fit the FAB criteria. Recently, genetic features (cytogenetic and molecular genetic) as well as history of myelodysplasia and prior therapy have been shown to have a significant impact on the clinical behavior of acute leukemias. The major limitations in the FAB classification of acute leukemias are the incorporation of these genetic and clinical features with morphology, since these features do not always correlate with the FAB categories. In 2001 World Health Organization (WHO) proposed a classification in which genetic information was incorporated with morphologic, cytochemical, immunophenotypic, and clinical information into diagnostic algorithms for the acute leukemias [8]. The recently published WHO 2008 classification expanded the number of entities with recurrent chromosomal translocations and included provisional entities [9]. Most hematopathologists continue to use both systems.
Acute Lymphoblastic Leukemias
ALL is one of the most common malignancies observed in the pediatric age group. It derives from the clonal proliferation of lymphoid progenitors in the bone marrow. ALL represents about 80 % of cases of acute leukemia in children but only 20 % in adults. The consequence of bone marrow infiltration is various cytopenias in the peripheral blood and is associated with the appearance of peripheral blast cells. In some instances, leukemic cells are not seen in the peripheral blood. Thus examination of bone marrow is usually necessary to confirm the diagnosis. Since the CNS is infiltrated at diagnosis in 5 % of patients, examination of the cerebrospinal fluid (CSF) by cytocentrifuge is also necessary.
ALL is a heterogeneous disease with several morphologic subtypes including mature B-ALL that are treated differently [10–12]. If a patient is correctly classified into a specific risk group, the cure rate can exceed 80 % [12, 13]. Therefore, subtype classification is very important in ALL diagnosis.
Morphologic Classification of Acute Lymphocytic Leukemia
The current classification of ALL was proposed by the FAB group in 1976 [6]. The FAB classification was based on the observation that approximately 80 % of children had a uniform population of small blast calls with high nuclear/cytoplasmic (N/C) ratio, a small inconspicuous nucleolus, and a regular nucleus These were designated L1 (Fig. 16.4). In a few patients the cells were described as more heterogeneous; more than 20 % had large cells with irregular nuclear membranes, one or more nucleoli, more cytoplasm, and lower N/C ratio. This subtype was called L2 (Fig. 16.5). A rare type seen in approximately equal percentages in children and adults (about 3–5 %), referred to as L3, is morphologically identical to the cell characteristics of Burkitt’s lymphoma. The cells are large and homogeneous with round to oval nuclei. The chromatin is finely dispersed with prominent nucleoli. The cytoplasm is intensely basophilic with or without vacuoles (Fig. 16.6). Of the three types described, only L3 is constantly associated with surface immunoglobulin (sIg) (mature B-ALL) [14]. In addition, an identical nonrandom chromosomal abnormality t(8;14) translocation has been described in both Burkitt’s lymphoma and ALL L3 [15]. In about 50 % of patients CD10 (CALLA) positivity has been reported along with the sIgs [16]. In rare instances, terminal deoxynucleodityl transferase-positive (TdT+) ALL with sIgs has been reported [17]. These cases do not have the typical chromosomal translocation or the L3 morphology.
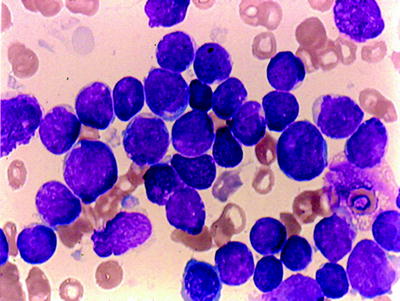
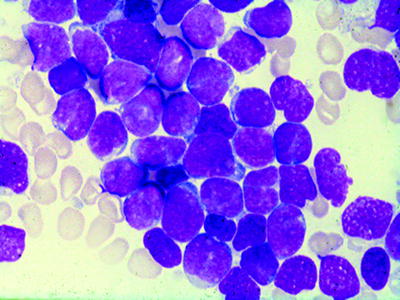
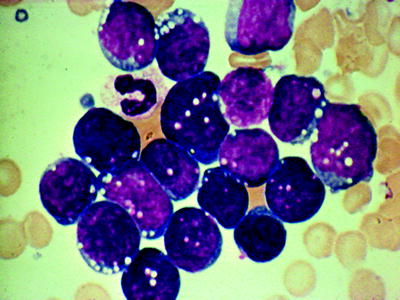
Fig. 16.4
Acute lymphocytic leukemia (ALL) (FABL1): agranular blasts with high nuclear/cytoplasmic ratio; smooth nuclear membrane; rare nucleoli. WG stain
Fig. 16.5
ALL (FABL2): More cytoplasm than L1 with prominent nucleoli. WG stain
Fig. 16.6
ALL (FABL3/Burkitt type): Blasts with oval nuclei, finely dispersed chromatin, basophilic cytoplasm with vacuoles. WG stain
Despite the accurate definition of these subtypes, observers registered discordance about their appreciation. The original FAB proposal was later modified in 1981 [18], eliminating some of the discordance among the observers and improving clinical correlations. The new scoring system was focused on three essential features of the blast cell population: N/C ratio, nucleoli, and irregular nuclear membrane. Cell size was another component when less than 50 % of leukemic cells were larger than twice the size of small lymphocytes. A simplified scoring system is given in Table 16.1. In this system, the sum may vary from +2 to −4. A total score of 0 to +2 defines L1, while a score of −1 to −4 establishes L2. With this revised system, concordance rates were close to 85 % in clinical studies [19].
Table 16.1
Simplified scoring system for FAB L1 and L2
Criteria | Score |
|---|---|
High N/C ratio ≥75 % of cells | + |
Low N/C ratio ≥25 % of cells | – |
0–1 Nucleoli (small) | + |
Irregular nuclear membrane ≥25 % of cells | – |
Large cells ≥50 % of cells | – |
Scoring range: +2 to −4 (L1:0−+2; L2: −1−4) | – |
Overall, L1 morphology is associated with a better event-free survival and induction rate than L2 [20], but this fact must be tempered because approximately 85 % of the L1 cases observed are in the pediatric age group. By contrast, L3 morphology was the worst prognosis.
The WHO 2008 no longer emphasizes the separation of L1 and L2.
Cytochemistry
By standard definition, the peroxidase reaction is totally negative in leukemic lymphoblasts. SBB stain is also negative. However, it has been suggested that SBB is less specific than MPO because positive reactions have been reported in ALL [21]. It should be remembered that in case of negative MPO or SBB reaction, megakaryocytic, erythroid, some monocytic, and AML M0 subtype as well as lymphoid lineage may be involved.
The PAS reaction is useful in supporting the diagnosis of ALL (Table 16.2), but its importance has declined, as immunophenotyping has become more important for this purpose. Although block-like PAS positivity is considered characteristic of ALL, only 15 % of cases have this type PAS reaction (Fig. 16.1) [6]. This pattern of PAS positivity correlates with the immunologic phenotype which is more common in B-lineage cases. The presence of both vacuolated blasts and PAS positivity correlates with reactivity of CD10 antigen [22]. It should be noted that block-like PAS positivity can be seen in monoblasts in AML M5 and in erythroblasts in AML M6 [23]. Moreover, fine granular activity in blocks of PAS material can be identified in all AML cell types (Fig. 16.7). PAS stain is therefore a poor discriminator of various cell types, but it may be useful as a part of battery of stains, particularly when many of the reactions are negative or nonspecific. A strong focal (paranuclear) acid phosphatase (ACP) activity or a strong focal ANAE activity greater than 75 % of blasts is characteristic of T-ALL [24]. However, it should be remembered that a localized ACP and ANAE reaction is also a feature of rare forms of AML: M7 and M6 [25, 26].
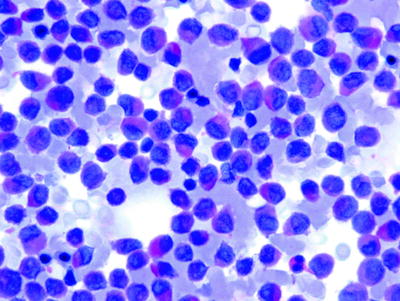
Table 16.2
Reaction of acute leukemic cells to cytochemical stains
Stain | AML (M1–M3) | AMML (M4) | AMoL (M5) | ALL (L1–L2) |
|---|---|---|---|---|
PAS | ± | + | ++ | ++ |
Acid phosphatase | – | ± | ++ | +++ |
Peroxidase | + | + | ± | – |
Chloroacetate esterase | + | + | – | – |
α-Naphthyl acetate esterase | ± | + | ++ | ++++ |
Fig. 16.7
AML: PAS reaction. Note the blush background
Immunohistochemistry
Indirect immunoperoxidase and indirect alkaline phosphatase anti-alkaline phosphatase (APAAP) techniques are applied to fixed cells in blood and bone marrow smears, cytospin preparations [27] or to paraffin-embedded material. These methods permit the detection of surface antigens (i.e., CD19, CD20, CD3, CD1a, and CD10), cytoplasmic antigens (CD79a, MPO, or cytoplasmic μ chain), and nuclear antigens (e.g., TdT and PAX5) [28].
Genetics of Acute Lymphoblastic Leukemia/Lymphoma
Acute lymphoblastic leukemia/lymphoma is relatively homogeneous at morphologic level. However, significant heterogeneity is seen at the genetic level. These genetic lesions define disease subsets with distinct biology and response to treatment and are used in the risk stratification schemas for current protocols [9, 29]. The six common subtypes of ALL are T-ALL, B lymphoblastic leukemia/lymphoma with t(9;22)(BCR-ABL), B lymphoblastic leukemia/lymphoma with t(4;11)MLL-rearrangement, B lymphoblastic leukemia/lymphoma with t(12;21)(TEL-AML1), B lymphoblastic leukemia/lymphoma with t(1;19)(E2A-PBX1), and B lymphoblastic leukemia/lymphoma with hyperdiploidy (with over 50 chromosomes).
The finding of prominent eosinophilia either T or B lymphoblastic leukemia/lymphoma must be considered for a rearrangement involving FGFR1. If none of the particular cytogenetic abnormalities (listed previously) are found in a case of B lymphoblastic leukemia/lymphoma, the designation of “B lymphoblastic leukemia/lymphoma not otherwise specified” is recommended. The term B-ALL should not be used for Burkitt’s lymphoma/leukemia, which is a neoplasm of mature B cells. Although abnormal karyotypes are frequently seen in T lymphoblastic leukemia/lymphoma, they are not clearly associated with unique biologic features. Therefore, T lymphoblastic leukemia/lymphoma is not usually subdivided further according to genetic abnormalities [9, 29–32].
Philadelphia Positive ALL
The true incidence of the Philadelphia chromosome in ALL is unknown. Cytogenetic studies suggest that it occurs in 2–4 % of children with ALL [33], but it is as high as 30 % in adults [34]. Thus, this chromosome appears to be the single most common genetic finding in adult ALL. Moreover, the presence of the Philadelphia chromosome identifies a subgroup of ALL with a poor prognosis [35]. Most bcr-abl-positive childhood cases express p190 fusion protein, whereas in adults, approximately 50 % of the cases express p210 kDa fusion protein (more likely in chronic granulocytic leukemia). It has been suggested that Philadelphia positive-bcr-positive ALL cases expressing p210 may be CML in lymphoid blast crisis; bcr-negative cases expressing variant p190 may be true de novo ALL [36]. However, no clinical data have been reported to substantiate this claim. One characteristic of Philadelphia-positive ALL is the expression of both myeloid and lymphoid antigens [37]. This feature supports that the cell origin is more immature than the other B-ALL cases [38]. There are no unique morphologic or cytochemical features that distinguish Philadelphia chromosome-positive ALL from other types of ALL.
Acute Myeloid Leukemias
AMLs result from a neoplastic transformation of a single pluripotential hematopoietic stem cell. Evidence to support this statement is morphologic [39], immunologic [40], and chromosomal [41] as well as from in vitro cell culture studies. These studies have demonstrated trilineage dysplastic features or lymphoid antigen expressions in AML, monocytic, and erythroid precursors in cultures and elaboration of colony-stimulating factors (CSFs) from monocytes.
Four major types of AML have long been recognized: acute myeloid, acute myelomonocytic, acute monocytic, and acute erythroleukemia. Despite these subtype definitions, Auer rods are the only consistent neoplastic marker demonstrating that a blast is myeloid and of leukemic origin. Auer rods are abnormal azurophilic crystalline-like granules that represent the coalescence of primary lysosomal granules of myeloid precursors as documented by cytochemical and ultrastructural studies [42, 43]. We have demonstrated that the addition of a peroxidase or another specific stain for primary granules increases the percentage of positive blast cells that contain Auer rods from 21 to 60 %. The clinical relevance of these observations is uncertain at the present time.
Morphologic Classification
The minimal requirements for morphologic specification of each case of AML are well-prepared peripheral blood and bone marrow smears prepared with a Romanowsky stain such as a Wright-Giemsa stain. To prove lineage, some routine cytochemical stains can be applied. These techniques are discussed next.
A uniform reproducible subtype classification in AML is necessary for several reasons; first, it permits comparison among various therapeutic regimens from different groups and from program to program within the same institution or cooperative group. Second, it allows for the potential identification of different clinical features and laboratory aspects that may be unique to certain subtypes. Finally with the increasing sophistication of chromosomal abnormalities in the AML, accurate description of myeloid subsets may permit a meaningful association of nonrandom rearrangements and translocations.
Morphologic classification of AML was proposed by the FAB Cooperative Group in 1976 [6] based on the morphologic and cytochemical features of the blast cells. The original proposal was revised and expanded in 1985 [44]. This classification is widely accepted and has been incorporated with immunologic and cytogenetic results in numerous studies (Table 16.3).
Table 16.3
Proposed WHO classification of acute myeloid leukemias (AML)
Acute myeloid leukemias with recurrent cytogenetic translocations |
AML with t(8;21)(q22;q22), AML 1/ETO |
Acute promyelocytic leukemia [AML with t(15;17)(q22;q11-12) and variants] |
PML/RARα |
AML with abnormal marrow eosinophils [inv(16)(p13q22) or t(16;16)(p13;q11)], |
CBFb/MYH11 |
AML with 11q23 (MLL) abnormalities |
Acute myeloid leukemia with multilineage dysplasia |
With prior myelodysplastic syndrome |
Without prior myelodysplastic syndrome |
Therapy-related acute myeloid leukemia |
Alkylating-agent-related |
Epipodophyllotoxin related (some may be lymphoid) |
Acute myeloid leukemia not otherwise categorized |
AML without maturation (FAB M0) |
AML with minimal maturation (FAB M1) |
AML with maturation (FAB M2) |
Acute myelomonocytic leukemia (FAB M4) |
Acute monocytic leukemia (FAB M5) |
Acute erythroid leukemia (FAB M6) |
Acute megakaryocytic leukemia (FAB M7) |
Acute basophilic leukemia (FAB M2Baso) |
Acute biphenotypic leukemia |
The FAB classification accepts two types of blasts: type I (agranular) blasts are myeloblasts with open chromatin, distinct nucleoli, and immature cytoplasm without granules; type II (granular) blasts are similar to type I, except that they contain up to 20 delicate cytoplasmic azurophilic granules or Auer rods, or both (Fig. 16.8). Goasguen et al. [45] distinguished another type of blasts from promyelocytes, referred to as type III blasts, these lack major characteristics of the promyelocyte, especially the Golgi zone and eccentric nuclear location, but they have many azurophilic granules. Type III blasts are characteristically identified in AML M2 associated with t(8;21), in secondary AML and AML developing from MDS [46]. In practice, although FAB type I and type II blasts can generally be distinguished from each other it has proved difficult to distinguish FAB type II blasts from type III blasts. In addition, the enumeration of abnormal promyelocytes in MDS or AML with multilineage dysplasia remains problematic and their separation from type II and type III blasts has remained imprecise. A group of international experts in morphology has provided guidelines to assist in this regard [47].
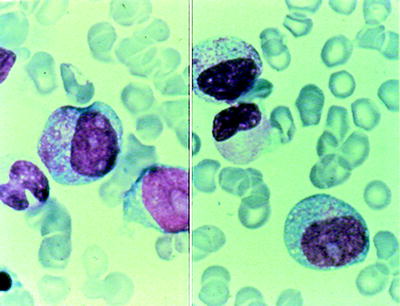
Fig. 16.8
Myeloblast morphology. Left panel: agranular blast and promyelocytes (note: Golgi zone); right panel: granular blast
Although FAB morphologic subtyping is inadequate as sole criteria in the classification of AML, it should continue to serve as a template from which to build other more clinically and relevant categories. FAB criteria identify one clinically significant type of AML, acute promyelocytic leukemia (M3); however, the remaining AML subtypes appear to be cytogenetically and immunologically heterogeneous group of diseases. Cytogenetic evaluation of AML identifies numerous prognostically significant types of genetic abnormalities; however, these do not precisely correlate with FAB categories. The 2001 World Health Organization (WHO) classification was proposed in an attempt to define more biologically homogeneous entities that have clinical relevance. As it relates to AML this includes (1) chromosomal abnormalities associated with recurrent translocations, i.e., t(15;17), t(8;21), inv(16) that are commonly seen in children and young adults, (2) Chromosomal abnormalities similar to those which are associated with myelodysplastic syndromes (MDS) (complex chromosomal abnormalities mainly loss of genetic material). These leukemias are associated with multilineage dysplasia of bone marrow cells, (3) AML cases without having chromosomal abnormalities or multilineage dysplasia. Later studies have validated this system [48].
The 2008 WHO classification expanded the number of entities with recurrent chromosomal translocations and included two provisional entities characterized by gene mutations, AML with cytoplasmic/mutated NPM1 and AML with CEBPA mutations [49, 50]. Therapy-related AML remains a category with important clinical and biological features. The diagnosis of myelodysplastic syndrome (MDS)-related AML has been further refined with specification of a history of prior MDS, presence of severe multilineage dysplasia, and MDS-associated cytogenetic abnormalities [51]. Myeloid proliferations of Down syndrome are separately classified [52]. It also retains FAB-like terminology for a morphologic classification of AML not otherwise specified (AML-NOS) [53].
In the WHO scheme, a myeloid neoplasm with 20 % or more blasts in the peripheral blood or bone marrow is considered to be AML [8]. The blast count should be obtained from at least a 200-cell count of all nucleated cells in the peripheral blood and a 500-cell count of all nucleated cells in the bone marrow. In certain cases associated with specific genetic abnormalities inv (16), t(8;21), t(16;16) or t(15;17), the diagnosis of AML is made regardless of the blast count in the peripheral blood or bone marrow. Similarly, the presence of granulocytic sarcoma is diagnostic of AML even if the blast count is less than 20 % in the blood or bone marrow [54].
Cytochemistry
Myeloperoxidase and Sudan Black B
An essential cytochemical stain is one that demonstrates the presence of MPO, an enzyme restricted to the primary granules of granulocytes and monocytes [7]. This enzyme can be identified by performing a benzidine-based peroxidase reaction or SBB staining or by diaminobenzidine reaction. The benzidine method is the best available method against which other methods should be compared. However, because of the concern for its potential carcinogenicity, it should not be adopted as the reference method [55].
The SBB reaction, a general stain for intracellular lipids, can be used in the same way. This stain is present in both primary and secondary granules. Sudanophilia and MPO activity are closely parallel [56].
Specific and Nonspecific Esterases
The chloroacetate esterase reaction can also be used to identify myeloid lineage. Although it is less sensitive than either MPO or SBB in detecting myeloid differentiation, it can be useful, when combined with ANAE in double esterase stain, in confirming a monocytic component. Because ANAE is positive both in neutrophils and in monocytes, inhibition by NAF in monocytes should be used to identify these cells [55, 56].
Periodic Acid-Schiff Reaction
All glycogen-containing cells as well as neutrophils and granulocytes at all stages of maturation stain with this reaction. Our experience in more than 200 patients has shown that fine granular PAS reaction can be identified in all AML subtypes, ranging from 60 % in AML (M1–M3) and 80 % in AML M5. Moreover, the latter often demonstrates a block-like reaction pattern characteristic of ALL (Bennett J.M., unpublished observation). The block-like pattern of PAS reactivity is seen in erythroid precursors of M6.
Electron Microscopy
Electron microscopy (EM) is used to demonstrate either MPO or platelet peroxidase (PPO). These ulltrastructural studies are useful in the confirmation of myeloid (MO) or megakaryocytic (M7) origin of blasts. MPO can be demonstrated by electron microscopy in blast cells that previously had negative results (less than 3 % positive blasts) by conventional criteria. It is also useful to define the myeloid component in biphenothpic leukemias [57, 58]. In megakaryoblasts, the PPO reaction is localized exclusively on the nuclear membrane and the endoplasmic reticulum, whereas in myeloblasts it occurs in the Golgi area and cytoplasmic granules.
Immunohistochemistry
Cytospin preparations or direct marrow or peripheral blood smears as well as bone marrow paraffin sections are used to demonstrate the presence of specific lineage antigens using a secondary technique (e.g., PAP, APAAP) to show antibody fixation [27]. Its advantage includes identification of morphologic features, the possibility of retrospective analysis and in situation peripheral blood or marrow smears are inadequate for study such as bone marrow fibrosis or hypocellular acute myeloid leukemia. Paraffin-reactive antibodies, i.e., CD34, MPO, CD68, CD117, CD41, CD61, Glycophophorin A, and Factor VIII-related antigen, can successfully be used to subclassify AML [59].
Acute Myeloid Leukemias with Recurrent Cytogenetic Abnormalities
All patients morphologically diagnosed with AML must have bone marrow cytogenetic studies performed. Molecular studies (FLT3, KIT, NPM1, and CEBPA) are an integral part of the WHO classification for AML lacking one of the recognized cytogenetic abnormalities. The revision of the WHO classification is recommended to incorporate recently described genetic aberrations into classification and define, biologically relevant, homogeneous entities not only based on the prognostic value of a genetic abnormality but also on morphologic, clinical, and phenotypic properties [54]. The contribution of cytogenetics to the classification of AML and acute myeloid leukemias with recurrent cytogenetic abnormalities are briefly discussed as follows.
Acute Myeloid Leukemia with t(8;21)(q22;q22);RUNX1-RUNX1T1
AML with t(8;21) is common in children and adults accounting for approximately 7–12 % of AML overall and 20–25 % of AML M2. Myeloid sarcomas may be present at presentation. Cases with t(8;21) should be diagnosed as AML regardless of a blast percentage in the bone marrow. The translocation results in a fusion product involving the RUNX1 gene (also known as core-binding factor alpha or AML1) on chromosome 21 and the RUNX1T1 (also known as ETO) gene on chromosome 8 [60]. Cases of AML with t(8;21) are associated with characteristic morphologic features [61]. Blasts are large cells with a distinctive nucleolus. The cytoplasm contains abnormal heavy azurophilic granulations (type III blasts) with tiny needle-like Auer rods (Fig. 16.9). The maturing neutrophils are commonly dysplastic. However, dysplasia of other cell lines is uncommon. Background eosinophilia is variably present. Blasts may express B-lineage antigens such as CD19, CD79a, PAX5, and TdT. These findings do not indicate the presence of mixed phenotype leukemia in the presence of t(8;21) [62]. Acute myeloid leukemia with t(8;21) is usually associated with favorable prognosis in children and adults [63]. Mutations of KIT occur in 20–30 % of cases [64]. Most published reports indicate a higher relapse rate and lower overall survival when mutated KIT is present [64, 65]. The presence of monosomy 7 as an additional cytogenetic abnormality may adversely impact prognosis [66]. Quantitative PCR measuring of RUNX1-RUNX1T1 transcripts is useful for monitoring minimal residual disease.
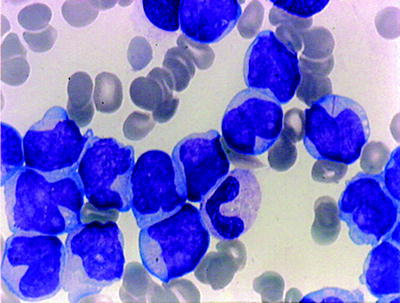
Fig. 16.9
AML –t(8;21). Note thin Auer rod in a myeloblast
Acute Promyelocytic Leukemia with t(15;17)/q22;q12, PML-RARA
APL is a distinct class of AML characterized by specific morphologic, cytogenetic, and clinical features. In most series, M3 accounts for about 10 % of cases in adults and for 4–8 % in children [67].
The highly specific t(15;17) translocation is present in at least 90 % of all APL cases. Using molecular techniques, virtually 100 % of APL cases have t(15;17). This translocation fuses the PML gene of the chromosome 15 to the retinoic acid receptor-α (RARA) gene, producing chimeric PML/RARA gene [68–70]. Cytogenetic analysis, FISH, or RT-PCR is necessary for genetic confirmation of the PML–RARA fusion. Detection of this abnormality is diagnostic of APL regardless of the blast count. The involvement of RARA at the translocation breakpoints may explain the clinical response to all-trans-retinoic acid (ATRA) [71, 72].
Typical APL is characterized by the presence of hypergranular blast cells, which have the appearance of abnormal promyelocytes. The cytoplasm is dense with coarse dark staining granules that often obscure the nucleus. The nuclear border is generally irregular and has a folded or reniform appearance (Fig. 16.10). Cells contain bundles of delicate needle-like Auer rods, giving a meshwork appearance (so called Faggots), found in some of the leukemic blasts. These abnormal promyelocytes are strongly positive for the MPO or SBB reactions. In contrast to other AML subtypes, dysplastic features of the myeloid series associated with leukemic proliferation are not seen. In adult patients with typical APL that achieve a CR, the prognosis is better than for any category of AML.
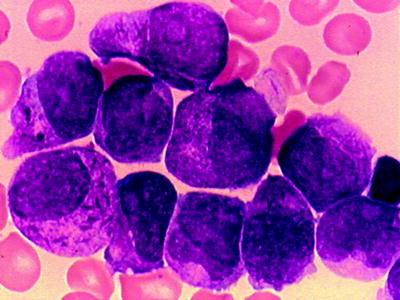
Fig. 16.10
APL (FABM3). Hypergranular promyelocytes with bundles of Auer rods
The FAB classification recognizes a variant of APL (FAB M3v) [73], characterized by bilobate cells or by cells with reniform nucleus and cytoplasm with minimal or no granulations (Fig. 16.11). Typical M3 cells are infrequent in the bone marrow. Hypogranular promyelocytes are strongly positive for the MPO or SBB, as in typical APL. The immunophenotype of M3v is identical to that described for the typical APL (expressing of myeloid antigens in the absence of HLA-DR reactivity) [74]. Special attention should be given to differentiate these cases from M4 and M5b subtypes. The distinction is important because of the well-known association of M3 with disseminated intravascular coagulation (DIC) [1]. The DIC syndrome is present equally in the M3v. In adults, the hypogranular or “microgranular” variant constitutes 25 % of APL cases [75]. It may also occur in children [76]. The hematological characteristic of M3v at onset, in addition to DIC, is hyperleukocytosis (>20,000/mm3). M3v has been associated with shorter CR and poorer overall survival, reflecting early hemorrhagic deaths [75, 76].
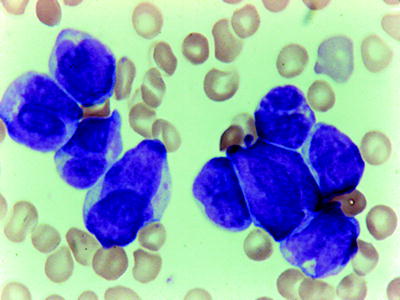
Fig. 16.11
APL-variant (FABM3v). Only occasional cells have granules but prominent bilobed and reniform nuclei. WG stain
FLT3 mutations are common in APL with the majority being internal tandem duplication (ITD) mutations. These mutations are strongly associated with the hypogranular subtype with hyperleukocytosis [77].
A small numbers of cases, often with morphologic features resembling APL, show variant translocations involving RARA gene on chromosome 17, but not the PML gene on chromosome 15, such as t(11;17) (ZBTB16-RARA), t(5;17)(NPM-1-RARA), and t(17;17)(STAT5B-RARA) [78]. Patients with variant RARA translocations often experience DIC. Cases with t(11;17) shows morphologic differences in which Auer rods are usually absent and pelgeroid neutrophils may be seen. Variant cases are important to recognize since some variants do not respond to ATRA [79].
Acute Myeloid Leukemia with inv(16)(p13.1q22) or t(16;16)p13.1;q22;CBFB-MYH11
AML with chromosome 16 abnormalities comprises 10 % of adult and 6 % of childhood AML. The inv(16) and the t(16;16) both result in the fusion of the beta subunit of core binding factor (CBFB) gene at 16q22 to the gene encoding smooth muscle myosin heavy chain (MYH11) at 16p13.1 [80]. The presence of this genetic abnormality is diagnostic of AML regardless of the blast count [49]. In these cases, in addition to characteristic morphologic features of acute myelomonocytic leukemia, the bone marrow shows a variable number of abnormal eosinophil components (AML-M4Eo) (Fig. 16.12). Eosinophils account for 5 % or more of nonerithroid cells. The eosinophils are abnormal; in addition to the specific eosinophilic granules, they have large basophilic granules and demonstrate chloroacetate esterase and PAS (coarse granules) positivity. Often the nuclei have pseudo-Pelger features. At least 3 % of the blasts show myeloperoxidase (MPO) or Sudan black B reactivity. The monoblasts and promonocytes usually show NSE or ANAE reactivity [81]. The incidence of extramedullary disease is higher than for most types of AML, with a high incidence of CNS relapse. Patients with chromosome 16 abnormalities tend to respond to chemotherapy better, experience relatively long remissions, and have a better prognosis [63, 82]. KIT mutations are present in 30 % of cases and negatively impact prognosis in older patients [64].
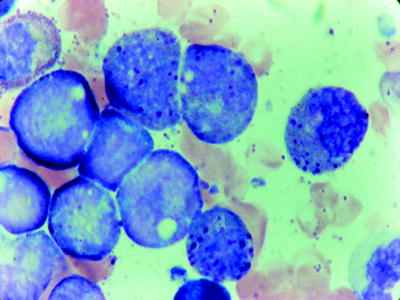
Fig. 16.12
AMML with eosinophils (FAB M4Eos). Note the magenta staining granules in the myelocyte. WG stain
Acute Myeloid Leukemia with t(9;11)(p22-q23); MLLT3-MLL
Translocations involving the MLL gene on chromosome 11q23 are seen in approximately 6 % of cases of AML and secondary leukemias that occur in patients treated with topoisomerase II inhibitors [83], acute lymphoblastic leukemia, and rarely in patients with MDS [84]. A number of different partners for the balanced 11q23 translocations have been identified. However, the WHO 2008 classification only recognizes t(9;11) (MLLT3-MLL) as a specific entity. This type of AML occurs at any age but is more common in children [85, 86]. Leukemic blasts containing 11q23 translocations generally have monocytic features and are subclassified as FAB M4 or M5 [87]. However, it is also detected in AML with or without maturation. Monoblasts and promonocytes show strong positive NSE reactions and often lack MPO reactivity. Extramedullary disease of the skin and gingiva and presentation with DIC has also been described.
Acute Myeloid Leukemia with t(6;9)(p23;q34);DEK-NUP214
The t(6;9) is detected in 0.7–1.8 % of AML that occurs in both children and adults.
In this entity there are no features specific to blast cells. The blasts may show occasional Auer rods and may exhibit monocytic features. Marrow and peripheral blood basophilia defined as more than 2 % is seen in half of the reported cases. Multilineage dysplasia may be evident in most of the cases [89, 90]. Blasts are MPO-positive. TdT may be positive in some cases. FLT3-ITD mutations are common in this type of AML occurring in 69 % of childhood and 78 % of adult cases [89, 90]. In all age groups AML with t(6;9)(p23;q34) has a poor prognosis.
Acute Myeloid Leukemia with inv (3)(q21q26.2) or t(3;3)(q21;q26.2);RPN1-EVI1
Acute myeloid leukemia with inv (3) or t(3;3) may represent de novo or may arise from MDS. It accounts for about 1–2 % of all AML cases and occurs most commonly in adults [85, 91]. Patients typically present with anemia and sometimes thrombocytosis [92, 93]. Dysplastic hypogranular neutrophils, pseudo-Pelger cells, dysplastic thrombocytes, and naked megakaryocyte nuclei associated with or without blast cells are noted as peripheral blood changes. Bone marrow blasts may show different morphologic features including FAB M1, M4, and M7 subtypes. MPO reactivity is often low. Multilineage dysplasia of bone marrow elements other than blast cells is a common finding. Dyserithropoesis and/or dysgranulopoiesis are frequent. Megakaryocytes may be normal or increased in number and usually have dysplastic features [93, 94]. Bone marrow cellularity and fibrosis are variable. A variety of abnormalities of the long arm of chromosome 3 occur in AML with inv (3) or t(3;3). High expression of the oncogene EVI1 at 3q26.2 is a poor prognostic indicator independent of 3q26 rearrangement. Secondary karyotypic abnormalities including monosomy 7, 5q deletions and complex karyotypes are present in most of the cases and associated with poor prognosis [94]. Patients with AML with inv (3) or t(3;3) typically have short survival [94, 95]. Some patients having these translocations may present less than 20 % blasts in the bone marrow and should be closely monitored for development of AML.
Acute Myeloid Leukemia (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
Acute myeloid leukemia with t(1;22) is a rare form of AML and restricted to infants and children younger than 3 years. It commonly occurs in infants without Down syndrome [49, 96]. The median age at diagnosis is 4 months. Majority of cases present with marked hepatosplenomegaly and/or osteolytic skeletal lesions with or without bone marrow involvement [97]. Patients also have anemia and thrombocytopenia with a moderately elevated white blood cell count. The morphology of the blast cells reveals that cells are very pleomorphic that may vary from very small with dense chromatin to somewhat larger with a fine reticulated nuclear chromatin and prominent nucleoli. Cytoplasmic blebs or actual platelet shedding may be found surrounding some blasts. The blasts are MPO or SBB-negative. Micromegakaryocytes are common, but dysplastic features of erythroid and granulocytic cells are not present. Bone marrow biopsy may show many clusters of micromegakaryoblasts, as well as more mature megakaryoblasts (Fig. 16.13). This is associated with an increase in reticulin formation and a corresponding decrease in normal hematopoietic precursors. Patients with t(1;22) and less than 20 % blasts on bone marrow aspiration should be correlated with the bone marrow biopsy. The presence of myeloid sarcoma is diagnostic of AML regardless of the marrow blast count. Megakaryoblasts express one or more of the platelet glycoproteins (CD41 and CD61). Cytoplasmic expression of these markers is more specific and sensitive than surface staining. The myeloid-associated markers CD13 and CD33 may be positive. CD36 is characteristically positive. CD34, MPO, HLA-DR, and TdT are negative [49]. In contrast to early studies that AML with t(1;22) is associated with poor prognosis, more recent studies suggested that they respond well to intensive AML therapy [98].
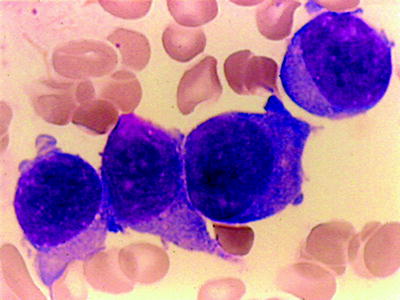
Fig. 16.13
Acute megakaryocytic leukemia. (FABM7). Large blasts with cytoplasmic projections. Immunostain for CD41 was positive. WG stain
AML with Gene Mutations
In addition to translocation and inversions, specific gene mutations also occur in AML. They include frequent mutations of fms-related tyrosine kinase 3(FLT3), nucleophosmin (NPM1), CEBPA, and KIT genes. Alone or in combination these mutations have recently been reported in AML patients with a normal karyotype. However, they may also be seen in patients with translocation and inversions as well [99]. Presence of these gene mutations has prognostic significance in the context of current therapies. Since these gene mutations do not define exclusive categories of AML and are present in many subtypes, they are provisional diagnostic categories in the WHO 2008 classification [49].
FLT3-ITD gene mutations may occur with any type of AML and MDS (20–40 % of cases) and their presence is associated with an adverse outcome [100]. KIT mutations have prognostic significance in cases of AML with t(8;21), inv(16), and t(16;16) (core binding factor leukemias), in which they are associated with a poor prognosis [64]. AML with mutated NPM1 is found in 50 % of adult and 20 % of pediatric AML patients with a normal karyotype [101, 102]. AML with mutated NPM1 in the absence of aFLT1-ITD mutation has a favorable prognosis [101, 103]. In adults, most of the NPM1-mutated cases show monocytic differentiation. Along with molecular techniques, paraffin sections are also used to show aberrant cytoplasmic expression of NPM by immunohistochemistry [102]. CEBPA mutations occur in 15–18 % of AML with normal karyotypes [104]. Presence of this mutation is associated with a favorable prognosis. There are no distinctive morphologic features.
Acute Myeloid Leukemia with Myelodysplastic-Related Changes
The category of AML with multilineage dysplasia from the WHO 2001 classification has been revised and named as “AML with myelodysplasia-related changes.” Patients are assigned to this group if they have 20 % or more blasts in the blood or marrow and (1) arise from previous myelodysplastic syndrome (MDS) or an myelodysplastic/myeloproliferative (MDS/MPN) neoplasm, (2) have specific MDS-related cytogenetic abnormalities, and/or (3) exhibit multilineage dysplasia (50 % or more of the cells in two or more myeloid lineages) [49]. The incorporation of these criteria now allows for a more prognostic and correct classification of cases with high blast count but minimal multilineage dysplasia and help to distinguish AML with myelodysplastic-related changes from AML FAB M6 and M7 in many cases. This category of AML occurs mainly in elderly patients and rare in children [105]. Myeloid sarcomas may be present at presentation and should be diagnosed as AML regardless of the bone marrow blast percentage [49].
To classify an AML as having myelodysplasia-related changes based on morphology, dysplasia must be present in at least 50 % of the cells in at least two myeloid cell lines. Morphologic dysplasias are characterized by neutrophils with hypogranular cytoplasm or hyposegmented nuclei; erythroblasts with megaloblastoid changes, nuclear irregularity, multinuclearity, and ringed sideroblasts; megakaryocytes with nonlobated or multiple nuclei; and micromegakaryocytes. Dysmegakaryopoiesis may be more easily recognized in paraffin sections [48, 106]. Some cases do not meet the criteria for a morphologic diagnosis. However, they are diagnosed as AML with myelodysplastic-related changes by the detection of MDS-related cytogenetic abnormalities and/or by a prior history of MDS or MDS/MPN. Some patients characterized by multilineage dysplasia have normal karyotype. These patients should be analyzed for FLT3, NPM1, and GEBPA mutations and if present should be included in the diagnosis [54]. Chromosome abnormalities are often involved in gain or loss of major segments of certain chromosomes with complex karyotypes that are similar to those found in MDS [82]. Balanced translocations are less common. The prognosis is unfavorable. Patients with MDS-associated cytogenetic abnormalities have a more consistently poor prognosis, whereas the absence of cytogenetic abnormalities in AML with myelodysplastic-related changes indicates a possible better prognosis [107].
Therapy-Related Myeloid Neoplasms
This category includes therapy-related AML, MDS, and MDS/MPN occurring as late complication of cytotoxic and/or radiation therapy administered for a neoplastic or non-neoplastic disorder [50]. Therapy-related myeloid neoplasms comprise 10 % AML and 20 % MDS cases. The incidence is increasing as more patients survive cancer.
Two subsets of therapy-related neoplasms are recognized. The longer latency cases (5–10 years after therapy) are associated with alkylating agents and ionizing radiation. This category is commonly associated with chromosomal losses, often of −5,−7 in a setting of complex karyotype [108, 109]. Shorter latency cases may arise 1–5 years after therapy and comprise 20–30 % of patients. These cases are usually associated with topoisomerase II inhibitor therapy and often have chromosomal translocations involving the MLL gene at 11q23 [109]. On the other hand, division of patients according to the type of the therapy is not practical since most patients have received polychemotherapy that includes both classes of drugs [110]. The bone marrow may be hypercellular, normocellular, or hypocellular and may have been associated with fibrosis. Blast counts are variable; approximately half of the t-MDS patients will have less than 5 % blasts [111].
In 20–30 % of cases, the first manifestation of the therapy-related myeloid neoplasm is overt leukemia without preceding myelodysplastic phase. Many cases fall in the FAB categories of AML M4 or M5, but cases of M2 also occur. Some cases may have karyotypic and morphologic changes identical to de novo AML, including acute promyelocytic leukemia. Such cases should be designated as t-AML with the appropriate cytogenetic abnormality indicated. Cases of lymphoblastic leukemia also occur in this group usually associated with t(4;11) [112]. The prognosis of therapy-related myeloid neoplasms is poor with overall survival reported at less than 10 %. Patients with MDS-related karyotypes have particularly dismal prognosis [113].
Acute Myeloid Leukemia, Not Otherwise Specified
Acute myeloid leukemia, not otherwise specified (AML,NOS) encompasses those cases that do not fulfill the criteria for any of the other previously described AML categories. AML, NOS accounts for 25–30 % of all cases. FAB terminology is retained for morphologic description of such cases. For diagnostic purposes it may still be important to recognize some particular disorders such as M7, M6, and acute panmyelosis with myelofibrosis (APMF). On the other hand, the epidemiology and clinical prognosis of these categories in the WHO classification is unknown because good and poor prognostic groups have been removed from the earlier FAB categories.
Acute Myeloblastic Leukemia without Cytologic Maturation (M0)
AML M0 is a rare form of acute myeloid leukemia consisting approximately less than 5 % of AMLs and may occur at any age. Morphologically, blasts are large with open chromatin and prominent single or multiple nucleoli. The N/C ratio is low. Cytoplasm is moderately basophilic. Azorophilic granules or Auer rods are not seen (Fig. 16.2). MPO or SBB-positive blasts are less than 3 % by light microscopy. Without immunophenotyping, AML M0 may be misdiagnosed as ALL based on the negative cytochemical reactions. So, it is important to stress that a diagnosis of AML M0 cannot be made on morphologic grounds alone and that always requires confirmation by immunologic techniques. Blast cells usually expressed CD13 and CD117 while expression of CD33 is found in approximately 60 % of cases. Since CD13 antigen is expressed in the cytoplasm of myeloblasts earlier than on the surface, CD13 should be tested by immunohistochemistry whenever the flow cytometry on the cell suspensions is negative. Moreover lymphoid differentiation antigens should be absent, except for TdT which is expressed in one-half of cases. Patients with the AML M0 subtype have a poorer response to combination therapy.
Acute Myeloblastic Leukemia with Minimal Differentiation (M1)
Poorly differentiated myeloblasts are the predominant nonerythroid cell type (type I and type II). Rarely, a case of M1 may present with majority of blasts type III. Auer rods may be present and consistent with the diagnosis (Fig. 16.14). More than 3 % of these blast cells are MPO or SBB-positive by conventional cytochemistry (Fig. 16.3). The low percentage of MPO-positive (3–10 %) M1 cases may constitute up to 25 % of all M1 cases [114]. In such cases, M1 should be differentiated from ALL L2, acute megakaryoblastic leukemia, and acute monoblastic leukemia without differentiation (M5a). Cytochemical stains and immunophenotyping using a classic panel (CD13, CD33, CD15, CD34, CD117) to confirm the myeloid nature of blasts are necessary for the differential diagnosis.
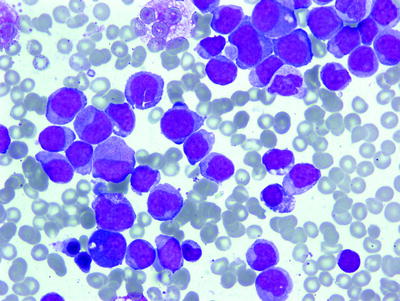
Fig. 16.14
AML (FABM1). Myeloblasts with <10 % maturation of granulocytes. WG stain
In AML M1, low MPO positivity cases were found to coexpress lymphoid and myeloid antigens. Moreover, discrepancy between immunologic MPO detection and light cytochemistry was found to be more frequent in patients with mixed immunophenotype than in patients without lymphoid markers [115].
Acute Myeloblastic Leukemia with Maturation (M2)
The defining characteristic that separates M2 from M1 is clear evidence of significant differentiation in all cells beyond the promyelocyte state (Fig. 16.15). The sum of the blast cells (type I + type II +, and type III) is greater than 20 % but less than 90 %, and generally there is a large predominance of type II blasts. Type III blasts are seen in AML M2 with t(8;21). Monocytic precursors cannot exceed 20 %. Increased numbers of eosinophilic precursors (M2Eo) as well as pseudo-Pelger-Hüet cells and hypogranular neutrophils may be seen. The MPO and SBB reactions are strongly positive. However, M2 cases with a partial MPO deficiency in the granulocytic precursors and mature granulocytes have been reported [116]. M2 may be associated with basophilia; this rare form must be separated from CGL blast crisis [117]. M2Eo should be separated from the more common M4Eo.
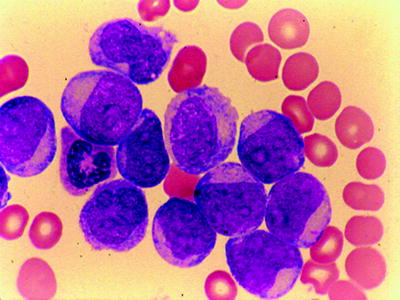
Fig. 16.15
AML (FABM2). Blasts with significant maturation of granulocytes at the promyelocyte and beyond. A single cell has an Auer rod. WG stain
Acute Myelomonocytic Leukemia (M4)
M4 accounts for 20–30 % of adult cases, depending on the application of esterase staining with fluoride inhibition. Its diagnosis and separation from M2 and M5 require assessment of both peripheral blood and bone marrow smears. In the bone marrow, both granulocytic and monocytic precursors exist in varying proportions (Fig. 16.16). The blast cells (types I, II, III) are greater than 20 % or more, but less than 80 %, of the nonerythroid cells. The marrow monocytic component must account for 20 % or more of the nonerythroid nucleated cells but cannot exceed 80 % (otherwise, the diagnosis is M5). Auer rods are occasionally present. When these findings are present with a peripheral blood monocytosis of 5,000/mm3 or greater, the diagnosis of M4 is not in question. If the peripheral monocytosis is less than 5,000/mm3, a diagnosis of M4 can still be made if the presence of a significant monocytic component (>20 %) is confirmed by cytochemical stain (Fig. 16.17) (e.g., double esterase reaction with NaF or other esterase combinations). Cytochemical stains are also necessary for confirming the diagnosis of M4 in the presence of hypogranular neutrophils that resemble monocytes. Dysplastic features involving granulocytic, erythroid, and megakaryocytic lineages can be identified in approximately 20 % of patients. Monocytic component can be identified by immunophenotyping using CD14, CD11c, CD11b, and CD68 in conjunction with other granulocyte-restricted antibodies.
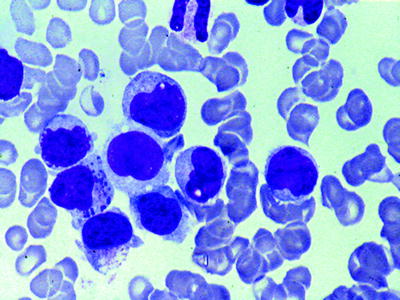
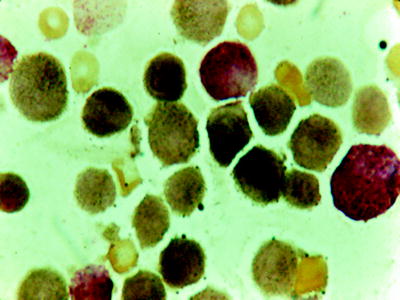
Fig. 16.16
AMML (FAB M4). Both myeloblasts and monocytic precursors are present. WG stain
Fig. 16.17
Double esterase reaction. Black product in monocytes and brick red color in granulocytes
Acute Monoblastic and Monocytic Leukemia (M5a and M5b)
AMoL accounts for about 10 % of the AML and is clonally expressed in cells committed to differentiation to monocytic pathway [118]. AMoL is divided into two morphologic subtypes: M5a and M5b.
M5a, or the poorly differentiated subtype, is frequently observed in children; it can be confused with the ALL L3 type. In the bone marrow, 80 % or more of all monocytic cells are monoblasts. Blast cells display abundant deep basophilic cytoplasm, which is often vacuolated and has no or few azurophilic granules and no Auer rods. The nuclei are round to oval, with one or more prominent nucleoli (Fig. 16.18). Since the peroxidase reaction may be negative in 40–50 % of such cases and the PAS reaction is often strongly positive with a block-like pattern, it is very useful to use nonspecific esterase (NSE) staining differential diagnosis. This stain will be strongly positive in more than 90 % of cases. Occasionally, the SBB reaction will be positive in the absence of a peroxidase reaction. Cells with monocytic differentiation can express at least two markers characteristic of monocyte differentiation such as CD14, CD4, CD11B, CD64, CD68, and CD36 on the cell surface.
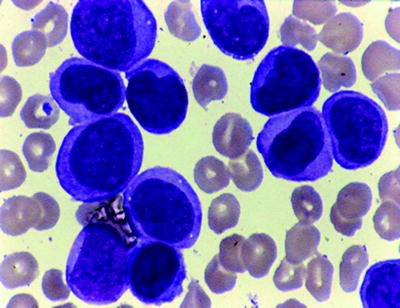
Fig. 16.18
AMOL (acute monocytic leukemia) (FABM5A). Large blasts with nuclear indentation without granules. WG stain
M5b, or the well-differentiated subtype, is defined by the presence of 20 % or more of abnormal cells being (promonocytes) with twisted or folded nuclei, gray-blue cytoplasm, and scattered azurophilic granules (Fig. 16.19). Rarely, a few cells will contain Auer rods. The percentage of mature monocytes is often much higher in the blood than in the bone marrow. Extramedullary masses (monocytic sarcomas), cutaneous or gingival infiltration, and CNS involvement are common. MPO and CEA are typically negative, but CD68 and CD168 are often positive in extramedullary myeloid (monoblastic) sarcomas.
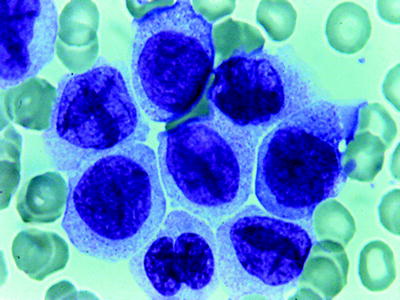
Fig. 16.19
AMOL (FABM5B). Promonocytes are apparent with occ. azurophil granules. WG stain
Acute Erythroid Leukemia (M6)
Erythroleukemia is a disease of adults and accounts for 5 % of AML. In 1976, the FAB cooperative group included erythroleukemia with the classification system of AML as M6 and agreed on a quantitative standard to be used in the diagnosis. The standards were revised in 1985 to the form used today [44]. The proposed criteria for the diagnosis of M6 are different from those used for the other types of AML. A minimum of 20 % blasts among the nonerythroid cells (NECs) and more than 50 % erythroid precursors with dysplastic features are necessary for diagnosis. However, erythroid leukemia of the WHO differs somewhat from FAB M6 [53]. One form (erythroid/myeloid) is a myeloblast proliferation that occurs in association with erythroid hyperplasia characterized by the presence of 50 % erythroid precursors in a total nucleated cell population in the bone marrow and 20 % myeloblasts among nonerythroid cells (NEC). All maturation stages of the erythroid precursors may be present (Fig. 16.20). They are dysplastic with megaloblastoid and/or multiple nuclei. The cytoplasm of the more immature erythroblast cells contains vacuoles. PAS may be positive either with a globular or a diffuse pattern. Ringed sideroblasts are present. Dysplastic changes of maturing neutrophils and megakaryocytes are common. Myeloblasts are poorly differentiated and contain occasional Auer rods. Erythroleukemia (erythroid/myeloid) must be distinguished from refractory anemia excess blasts (RAEB), and AML with myelodysplasia-related changes. Many of these cases are now precisely classified as AML with myelodysplasia-related changes when total marrow blasts are ≥20 % and multilineage dysplasia is present in more than 50 % of the cells of two or more lineages. When there are <20 % blasts and the erythroid precursors are ≥50 % of the cells, the differential count of NEC must be calculated. If blasts are ≥20 % of NEC the diagnosis is erythroleukemia (myeloid/erythroid); if ≤20, the diagnosis is MDS.
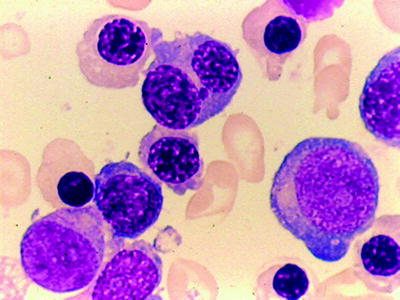
Fig. 16.20




Acute erythroleukemia (FABM6A). Basophilic erythroid precursors and maturing erythroid cells. Megaloblastic changes. One myeloblast with an Auer rod. WG stain
Related posts:



Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
