INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative disorder of pluripotent hematopoietic stem cells, characterized by the molecular BCR-ABL1 rearrangement, which drives a proliferative and survival advantage of the leukemic clone. About 30% to 50% of patients with CML are asymptomatic at diagnosis and are incidentally diagnosed during routine examination. Patients may also present with characteristic clinical findings secondary to large numbers of myeloid circulating progenitors, leading to splenomegaly, leukocytosis, or even isolated thrombocytosis. The landscape of CML has had a dramatic course, with a host of findings that have elucidated the biology and molecular pathology of the disease. The understanding of the molecular events led to the creation of the first targeted therapy—imatinib mesylate. Its impact on therapy and survival propelled CML as a model for modern molecular medicine in the fast-developing era of personalized targeted therapy.
EPIDEMIOLOGY AND ETIOLOGY
The incidence of CML is 1 to 2 cases per 100,000 adults with a slight male predominance and rising incidence with age, accounting for approximately 15% of newly diagnosed cases of leukemia in adults (1). Chronic myeloid leukemia is uncommon in children, with a median age at diagnosis of 67 years. There are no known hereditary, geographic, familial, or ethnic associations. No chemical or infectious associations have been established, although an increased risk has been linked with exposure to ionizing radiation (2).
In 2014, in the United States, an estimated 6,000 cases of CML were diagnosed. Since 2000, the year of introduction of imatinib, the annual mortality in CML has decreased from 10% to 20% to 1% to 2%. Consequently, the prevalence of CML in the United States, which was estimated at about 25,000 to 30,000 cases in 2000, has increased to an estimated 80,000 to 100,000+ cases in 2015 and will reach a plateau of about 180,000 cases by 2030 (3). The overall survival (OS) of patients over the recent decade has greatly improved. The exact mechanism of initiation of the defining molecular event in CML—Philadelphia (Ph) chromosome translocation—remains elusive.
BIOLOGY OF CHRONIC MYELOID LEUKEMIA
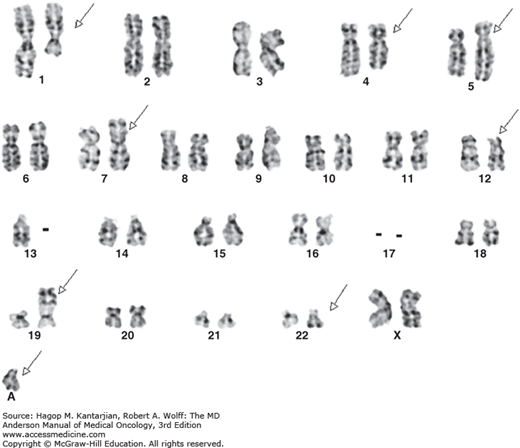
Chronic myeloid leukemia is defined by a unique cytogenetic and/or molecular abnormality—the Ph chromosome—originating in a pluripotent stem cell, with a balanced translocation between the long arms of chromosomes 9 and 22, t(9,22)(q34,q11.2) (4). Detectable by routine cytogenetics or by fluorescent in situ hybridization (FISH), the Ph chromosome is noted in 90% to 95% of patients with the clinical and laboratory features of CML. In the remaining 5% to 10% of patients, the molecular BCR-ABL1 rearrangement can be recognized with high-sensitivity reverse transcriptase polymerase chain reaction (RT-PCR). All remaining cases with unknown biology deemed as true Ph-negative CML or atypical CML carry a poor prognosis. The Ph chromosome joins the proto-oncogene c-abl from chromosome 9 to the breakpoint cluster region (BCR) gene in chromosome 22, generating a novel fusion BCR-ABL1 oncogene. Depending on the breakpoint region in BCR, several variants including those of 190, 210 (most common; 95% of Ph-positive cases), or 230 kDa molecular weight can be formed. The translation product is a chimeric protein with constitutively active tyrosine kinase activity that activates multiple downstream pathways, including PI3 kinase, NF-κB, JAK/STAT, RAS, RAF, ERK, MYC, and JNK.
Following the 2005 discovery of the JAK2 V617F mutation as the molecular abnormality common to polycythemia vera, essential thrombocytosis, and idiopathic myelofibrosis, CML has been segregated into a separate group of myeloproliferative neoplasms categorized as BCR-ABL1 positive/JAK2 V617F negative.
Roughly 10% to 15% of patients, typically at a more advanced stage of disease, experience clonal evolution with additional chromosomal aberrations including trisomy 8, monosomy 7, isochrome 17, a double Ph chromosome, or additional loss of material from 22q. Clonal evolution is considered a criterion of accelerated phase, particularly when it occurs during the course of the disease. Corresponding molecular alterations that follow these changes include deregulation of p53, RB1, C-MYC, and AML-EVI1.
CLINICAL FEATURES AND NATURAL HISTORY
There are three separate phases of CML: chronic phase (CP), intermediate or accelerated phase (AP), and terminal or blast phase (BP). Even though all three represent a stepwise progression of disease in terms of aggressiveness from CP to BP, the natural course of the disease may not progress from one to the other, nor will disease always include all three. The vast majority of patients are diagnosed as asymptomatic in the CP. Splenomegaly is the most consistent presenting sign, detected in 50% to 60% of cases. Previously defined criteria indicating progression from CP to AP include 15% or more blasts, 30% or more blasts plus promyelocytes, 20% or more basophils, platelets <100 × 109/L unrelated to therapy, or cytogenetic clonal evolution (5). Other criteria have been proposed (Table 4-1). Compared to CP, median survival in AP is significantly shorter, although a significant improvement in survival has been observed with the availability of tyrosine kinase inhibitors (TKIs), particularly among early responders (6).
| MDACC | IBMTR | WHO | |
|---|---|---|---|
| Blasts | ≥15% | ≥10% | 10-19a |
| Blasts+Pros | ≥30% | ≥20% | NA |
| Basophils | ≥20% | ≥20%b | ≥20% |
| Platelets (× 109/L) | <100 | Unresponsive ↑, persistent ↓ | <100 or >1000 unresponsive |
| Cytogenetics | CE | CE | CE not at diagnosis |
| WBC | NA | Difficult to control, or doubling <5 days | NA |
| Anemia | NA | Unresponsive | NA |
| Splenomegaly | NA | Increasing | NA |
| Other | NA | Chloromas, myelofibrosis | Megakaryocyte proliferation, fibrosis |
Most patients evolve into AP prior to BP, but 20% progress to BP without AP warning signals. Blast phase is considered an acute leukemia. It is defined by the presence of at least 30% blasts in the peripheral blood or bone marrow or the presence of extramedullary disease (chloroma or granulocytic sarcoma). Half of the patients in BP carry a myeloid phenotype, whereas the other half is split evenly between lymphoid and undifferentiated (7). Median survival in BP CML remains poor; a combination of TKI with chemotherapy followed by allogeneic stem-cell transplantation is recommended.
DIAGNOSIS AND CLINICAL WORKUP
Initial evaluation aims to elicit signs and/or symptoms of disease, whereas physical examination evaluates for any presence of organomegaly or extramedullary hematopoiesis. Diagnosing typical CML requires documentation of the Ph chromosome abnormality by routine cytogenetic evaluation (karyotype), FISH, or molecular studies (RT-PCR) (8). Bone marrow aspiration and biopsy are required, because they will not only confirm the diagnosis (eg, cytogenetic analysis), but also provide information needed for staging and classification. Although FISH, which relies on co-localization of large genomic probes specific to the BCR and ABL genes, or RT-PCR from peripheral blood can both confirm the presence of the Ph chromosome or the BCR-ABL1 rearrangement, only bone marrow cytogenetics can reveal additional chromosomal abnormalities that are important in the initial diagnosis and staging.
True Ph- and BCR-ABL1–negative patients are considered to have Ph-negative CML or chronic myelomonocytic leukemia. In a few instances, a patient may be diagnosed with Ph-positive acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL). It may be impossible to determine whether these cases represent de novo Ph-positive acute leukemias or a BP of a previously unrecognized CML.
LABORATORY FEATURES
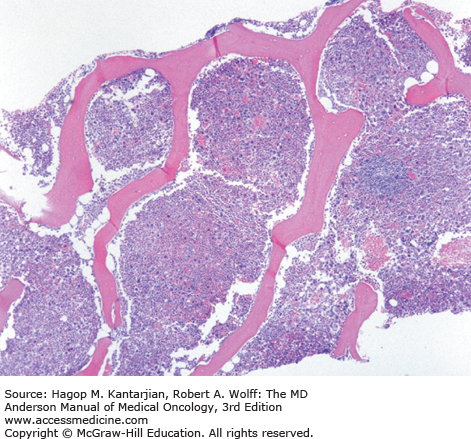
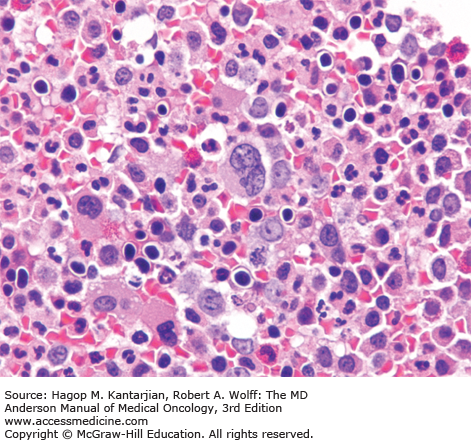
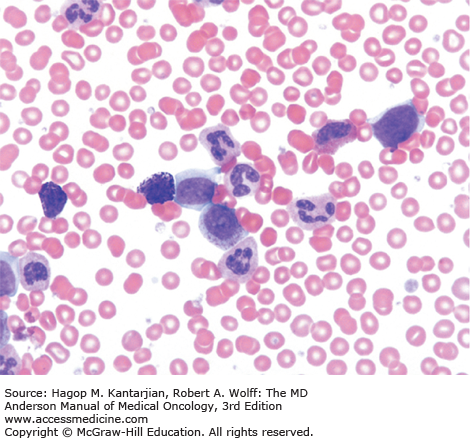
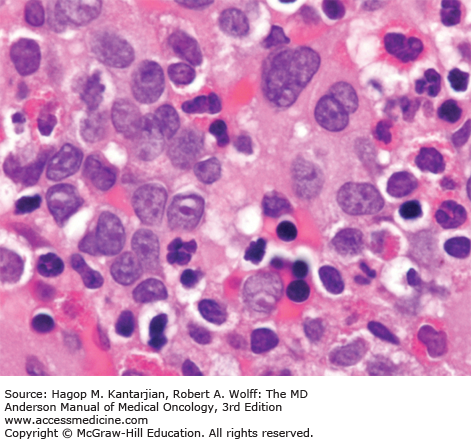
The most common laboratory finding in CP CML is leukocytosis, with myeloid cells in all stages of maturation seen in the peripheral blood. Frequently, there is an increase of basophils and eosinophils. The bone marrow is markedly hypercellular, with the myeloid-to-erythroid ratio significantly increased (Figs. 4-1,4-2,4-3,4-4,4-5) (9).
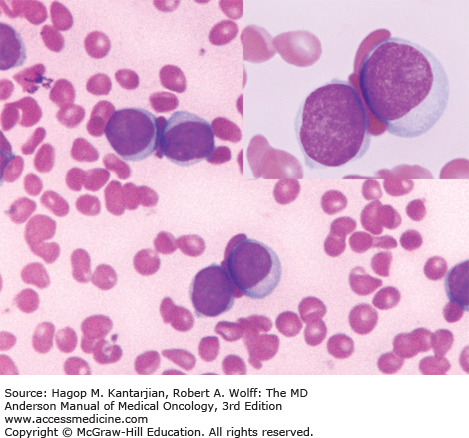
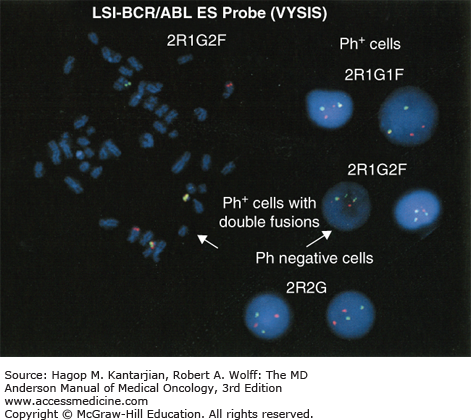
A marrow aspiration and chromosomal analysis are needed to identify any variances in marrow blasts or basophils and to identify the Ph chromosome and possible cytogenetic clonal evolution (Fig. 4-6). Fluorescent in situ hybridization and PCR should be done at diagnosis in all patients. FISH may identify the presence of the BCR-ABL1 rearrangement, even when the Ph chromosome is not found by conventional cytogenetic analysis. Furthermore, FISH can be performed on peripheral blood interphase cells (Fig. 4-7). Reverse transcriptase PCR testing amplifies the region around the splice junction between BCR and ABL and is highly sensitive for detecting CML, identifying the initial type of BCR-ABL1 transcript (important for follow-up), and especially quantifying minimal residual disease (MRD). In general, simultaneous peripheral blood and marrow quantitative PCRs have a high level of concordance, although false-positive and false-negative results can be seen (false-negative results due to poor-quality material or failure of the reaction; false-positive results due to contamination). Quantitative PCR assesses the amount of BCR-ABL1 transcripts and is universally performed. Table 4-2 presents a summary of the proposed evaluation at diagnosis and during follow-up for patients with CML.
| Status | Diagnostic Testing |
|---|---|
| At diagnosis |
|
| During therapy |
|
| |
| |
|
PROGNOSIS
As the newer generation agents became available, the prognostic significance of certain clinical characteristics changed. Achieving a complete cytogenetic response (CCyR) is the most important prognostic factor for long-term survival (10). It has become evident that achieving a cytogenetic response, particularly during the first 3 to 6 months of therapy, translates into the best probability of long-term outcome.
RESPONSE AND EVALUATION OF MINIMAL RESIDUAL DISEASE
Response to therapy is initially judged by measurement of hematologic response criteria. A complete hematologic response (CHR) is defined as normalization of white blood cell (WBC) counts to <10 × 109/L with a normal differential, platelet count <450 × 109/L, and disappearance of splenomegaly and other symptoms of CML. Patients who achieve a CHR are further classified according to the type of cytogenetic response attained (Table 4-3).
| Hematologic remission | |
| Complete | Normalization of peripheral counts and differential, and disappearance of all signs and symptoms of CML including splenomegaly |
| Cytogenetic remissiona | |
| Complete | 0% Ph-positive metaphases |
| Partial | 1%-35% Ph-positive metaphases |
| Minor | 36%-95% Ph-positive metaphases |
| None | >95% Ph-positive metaphases |
| (Complete and partial remissions together constitute major cytogenetic remissions, ie, 0%-34% Ph-positive metaphases) | |
| Molecular remissionb | |
| Complete | Undetectable BCR-ABL transcriptsc |
| Major | BCR-ABL/ABL ratio of <0.1% (International Scale) |
Cytogenetic responses are divided into complete, partial, and minor. Complete cytogenetic response corresponds to 0% of all metaphases remaining Ph positive; partial cytogenetic response is defined as 1% to 35% of metaphases being Ph positive; and minor cytogenetic response is defined as 35% to 95% Ph-positive metaphases. An analysis involving review of at least 20 metaphases is necessary to evaluate a cytogenetic response. Although FISH results correlate well with the karyotypic evaluation, cytogenetic response criteria have not been validated with FISH.
Molecular response is assessed by quantitative PCR (usually RT-PCR) in the peripheral blood or bone marrow (11). A major molecular response (MMR) is considered when the BCR-ABL1/ABL1 ratio is ≤0.1% on the international scale (IS). A complete molecular response (CMR) represents achievement of undetectable transcripts of BCR-ABL1 in an assay with a sensitivity of at least 4.5 logs. For correlative purposes, BCR-ABL1 transcript levels (IS) of ≤1% are equivalent to a CCyR; levels of ≤10% are equivalent to a partial cytogenetic response.
THERAPY
Three commercially available TKIs are approved for the frontline treatment of CML: imatinib, dasatinib, and nilotinib. Available guidelines support all three as viable frontline options for the initial management of CP CML.
Prior to the development of targeted therapy, CML was treated with busulfan or hydroxyurea for many years, with a poor prognosis and inability to delay disease progression. The introduction of interferon-alfa (IFN-α) improved survival in CML and resulted in CCyRs in 5% to 25% of patients with CP CML.
Imatinib mesylate (STI-571 or Gleevec) is a selective and potent competitive inhibitor of the adenosine triphosphate (ATP)–binding site of the Bcr-Abl oncoprotein, as well as c-kit, PDGFRα and PDGFRβ, and abl-related gene (ARG) (12). It is taken orally with 98% bioavailability and a half-life of 13 to 16 hours. It was first used in CML patients who developed resistance or intolerance to IFN-α and resulted in a CCyR of 60% and an estimated 5-year OS of 76% (13).
Based on these results, the large International Randomized Study of Interferon and STI571 (IRIS) trial evaluated imatinib versus IFN-α and low-dose cytarabine as frontline therapy in patients with newly diagnosed CP CML. Patients were randomized to receive imatinib 400 mg/d or INF-α plus low-dose subcutaneous cytarabine, which was standard of care at the time (10). After a median follow-up of 19 months, planned outcome analyses were significantly better in almost all categories for patients receiving imatinib. The rates of CCyR (74% vs 9%; P < .001) and freedom from progression to AP or BP at 12 months (99% vs 93%; P < .001) were improved. There was a high crossover rate to imatinib. The responses to imatinib were durable, as outlined in an 8-year follow-up of the original study (14). Estimated event-free survival was 81%, and OS was 93% when only CML-related deaths were considered. With an annual mortality of 2%, the estimated median survival of a newly diagnosed patient with CML may be in the range of 20 to 30 years.
A phase I imatinib study in patients who had failed prior IFN-α therapy established a clear relationship between dose and response (15). No significant responses were observed at doses <300 mg daily. An arbitrary dose of 400 mg daily for CP was selected in phase II studies, despite the lack of dose-limiting toxicity at doses up to 1,000 mg daily (maximum-tolerated dose was not defined). The Rationale and Insight for Gleevec High-Dose Therapy (RIGHT) trial studied imatinib 400 mg twice a day as initial therapy in 115 patients with newly diagnosed CML (16). The rate of CCyR was 85% at 12 months and 83% at 18 months, with corresponding rates of MMR of 54% at 12 months and 63% at 18 months.
These results led to a randomized phase III open-label study, the Tyrosine Kinase Inhibitor Optimization and Selectivity Trial (TOPS), comparing 400 and 800 mg of imatinib in 476 patients. The trial showed significant superiority for the 800-mg dose in terms of MMR rate at 3 months (3% vs 12%), 6 months (17% vs 34%), and 9 months (33% vs 45%) but not at 12 months (40% vs 46%) (17). A final update of the trial showed no significant difference in MMR rates at 24 months (52% vs 50%) (18). Another European study also reported no benefit with imatinib 800 mg compared to 400 mg in high-risk CML (CCyR rates were 64% and 58% and MMR rates were and 40% and 33%, respectively) (19).
The French SPIRIT study evaluated the impact of adding IFN or cytarabine in a randomized study where 636 patients with untreated CP CML received one of the following: imatinib 400 mg daily alone, imatinib 400 mg daily with cytarabine (20 mg/m2/d on days 15-28 of each 28-day cycle) or pegylated IFN-α-2a (90 μg weekly), or imatinib 600 mg daily alone (20). The rates of cytogenetic response at 12 months were similar among the four groups, whereas the rate of molecular response (a decrease in the ratio of BCR-ABL1/ABL1 of ≤0.01%) was significantly higher in patients receiving imatinib and pegylated IFN-α-2a compared with imatinib 400 mg alone arm (30% vs 14%, respectively; P = .001). This rate was also significantly higher in patients treated for more than 12 months compared to treatment lasting ≤12 months. However, this high rate of early and deep responses did not translate into long-term improvement due to the poor tolerance of pegylated IFN.
Imatinib 400 mg daily is the regimen of choice for newly diagnosed patients with CP CML, with an emphasis on maintaining adequate dose intensity with minimal treatment interruptions or dose reductions for the best outcome.
Imatinib is well tolerated, although adverse events not requiring treatment interruptions or decrease in dosing may occur in 30% to 40% of patients. A list of some of the most frequently encountered side effects and suggestions for management are included in Table 4-4. Any grade 3 or 4 toxicities related to imatinib require treatment interruption and resumption upon resolution of toxicity or its decrease to grade 1 or less. Subsequent dose should be reduced if recurring or long-lasting adverse effects are encountered, keeping in mind that doses below 300 mg daily are not recommended due to lack of adequate activity. Only 2% to 3% of patients exhibit true intolerance to imatinib and require permanent discontinuation. Early recognition and intervention targeting toxicities greatly reduce the need for unnecessary treatment interruptions and dose reductions.
| Adverse Events | Management |
|---|---|
| Nausea/vomiting | Take with food, fluids |
| Antiemetics | |
| Diarrhea | Loperamide |
| Diphenoxylate atropine | |
| Peripheral edema | Diuretics |
| Periorbital edema | Steroid-containing cream |
| Skin rash | Avoid sun exposure |
| Topical steroids | |
| Systemic steroids | |
| (Early intervention important) | |
| Muscle cramps | Tonic water or quinine |
| Electrolyte replacement as needed | |
| Calcium gluconate | |
| Arthralgia, bone pain | Nonsteroidal anti-inflammatory agents |
| Elevated transaminases (uncommon) | Hold therapy and monitor closely |
| Dose reduction upon resolution | |
| Myelosuppression | |
| Anemia | Treatment interruption/dose reduction usually not indicated |
| Erythropoietin or darbepoetin | |
| Neutropenia | Hold therapy if grade ≥3 (ie, ANC <1 × 109/L) |
| Restart at lower dose if recovery takes >2 weeks | |
| Consider G-CSF if recurrent/persistent, or sepsis | |
| Thrombocytopenia | Hold therapy if grade ≥3 (ie, platelets <50 × 109/L) |
| Restart at lower dose if recovery takes >2 weeks | |
| Consider IL-11 10 μg/kg 3-7 days/week |
Myelosuppression is common and frequently seen within the first 2 to 3 months of therapy. It is generally self-limited, and dose interruptions are not recommended unless grade 3 neutropenia or thrombocytopenia (ie, neutrophils <1 × 109/L, platelets <50 × 109/L) develops. Anemia alone usually does not require interruptions or dose adjustments. Treatment is restarted when counts recover above specified thresholds. Following treatment interruption, WBC should be monitored at least once weekly, and if recovery occurs within 2 weeks, treatment would be resumed with the same dose at which myelosuppression occurred. If recovery takes longer than 2 weeks, the dose could be reduced in increments (eg, from 800 to 600 mg, from 600 to 400 mg, or from 400 to 300 mg). Hematopoietic growth factors may be beneficial with recurrent or prolonged myelosuppression (eg, erythropoietin or darbepoetin and filgrastim).
Dasatinib (Sprycel) is an oral second-generation TKI that is a piperazinyl derivative. It has an excellent oral bioavailability and is 350 times more potent in vitro than imatinib (21,22) (in vitro sensitivity of different BCR-ABL1 mutants to different TKIs is presented in Table 4-5) (23). Dasatinib exhibits significant activity against most imatinib-resistant BCR-ABL1 mutations, with the exception of T315I, as well as a few others, including V299L and F317L (24). In contrast to imatinib, dasatinib binds to both the active and inactive conformations of BCR-ABL1 and also inhibits the Src family of kinases, which may be important in suppressing critical cell signaling pathways (25).
| BCR-ABL Mutant | Ponatinib | Imatinib | Nilotinib | Dasatinib | Bosutinib |
|---|---|---|---|---|---|
| Native | 3 | 201 | 15 | 2 | 71 |
| M244V | 3 | 287 | 12 | 2 | 147 |
| L248R | 8 | 10000 | 549 | 6 | 874 |
| L248V | 4 | 586 | 26 | 5 | 182 |
| G250E | 5 | 1087 | 41 | 4 | 85 |
| Y253H | 5 | 4908 | 179 | 3 | 40 |
| E255K | 6 | 2487 | 127 | 9 | 181 |
| E255V | 16 | 8322 | 784 | 11 | 214 |
| V299L | 4 | 295 | 24 | 16 | 1228 |
| T315A | 4 | 476 | 50 | 59 | 122 |
| T315I | 6 | 9773 | 8091 | 10000 | 4338 |
| F317C | 3 | 324 | 16 | 45 | 165 |
| F317I | 7 | 266 | 25 | 40 | 232 |
| F317L | 4 | 675 | 21 | 10 | 82 |
| F317V | 10 | 1023 | 26 | 104 | 1280 |
| M351T | 4 | 404 | 15 | 2 | 97 |
| E355A | 7 | 441 | 18 | 3 | 74 |
| F359C | 6 | 728 | 47 | 2 | 70 |
| F359I | 11 | 324 | 64 | 3 | 76 |
| F359V | 4 | 346 | 41 | 2 | 59 |
| H396R | 4 | 395 | 23 | 2 | 60 |
| E459K | 5 | 612 | 38 | 4 | 127 |
| Criteria Used to Classify Drug Potency | |||||
| Ponatinib | Imatinib | Nilotinib | Dasatinib | Bosutinib | |
| Effective Cave at recommended dose | 28a | 444 | 131 | 11 | 159 |
| IC50 <75% of Cave | <21 | <333 | <98 | <8 | <119 |
| IC50 75%-150% of Cave | 21-32 | 333-500 | 98-147 | 8-12 | 119-179 |
| IC50 150%-300% of Cave | 33-95 | 501-1499 | 148-442 | 13-37 | 180-537 |
| IC50 >300% of Cave | >95 | >1499 | >442 | >37 | >537 |
Following evaluation in the salvage setting after imatinib failure, dasatinib was assessed in the frontline setting. The DASISION trial was a phase III randomized study that compared imatinib 400 mg once daily to dasatinib 100 mg once daily in 519 patients with newly diagnosed CP CML (26). The primary end point was confirmed CCyR at 12 months. The dasatinib arm resulted in higher confirmed CCyR at 12 months (77% vs 66%; P = .007). The rates of molecular response were significantly higher with dasatinib (MMR, 76% vs 64%, P = .002; molecular response with a 4.5-log reduction in BCR-ABL transcripts from baseline [MR4.5], 42% and 33%, P
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


