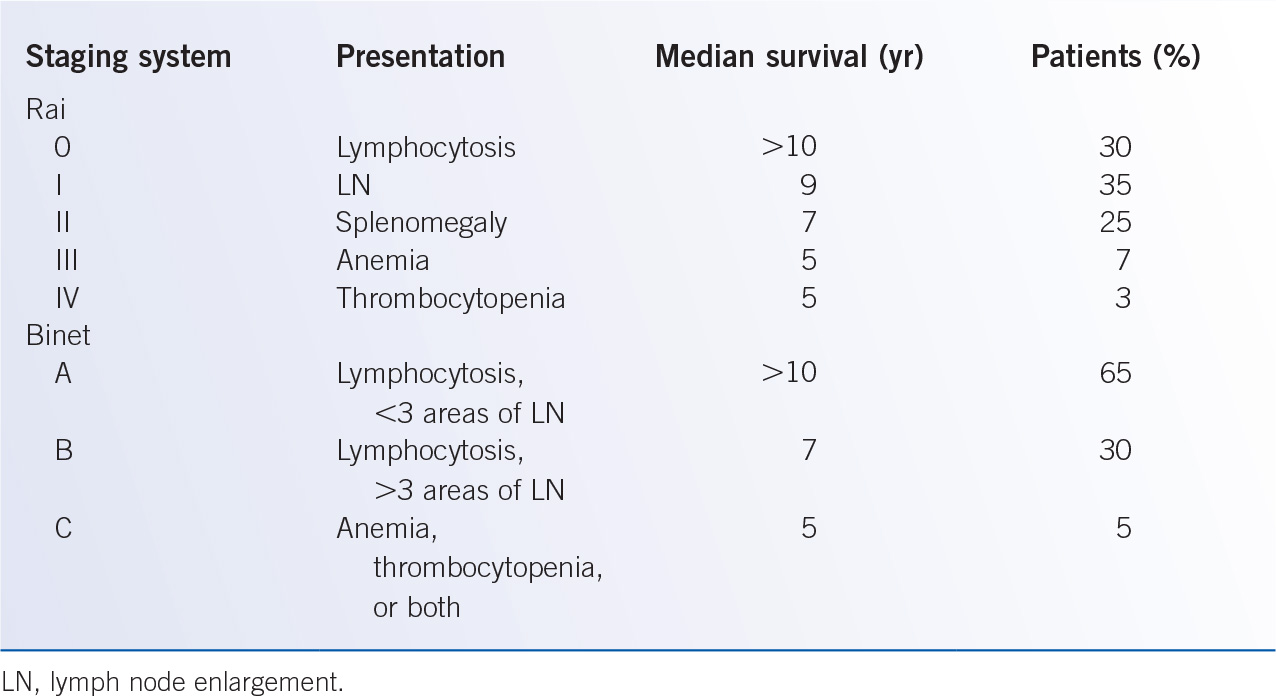
D. Staging and prognosis. The Rai and Binet clinical staging systems were described in the 1970s and provided prognostic information on survival based upon physical examination findings and blood counts (Table 28-3). These clinical staging systems have been extensively validated and are widely used in clinical practice to provide estimates of survival to patients. However, the clinical course of early stage patients is heterogeneous: some patients do not require therapy for many years, whereas others have rapid progression and poor responses to therapy. Further, these staging systems provide little information to help predict the clinical outcome of early stage patients or the response to therapy. Additional laboratory parameters have been identified as markers of tumor burden and independent poor prognostic factors including an elevated LDH, a lymphocyte doubling time of less than 12 months, and diffuse bone marrow infiltration pattern. Serum proteins have also been linked to poor prognosis, including elevated levels of thymidine kinase (TK), soluble CD23 (sCD23), and B2-microglobulin (B2M). Notably, patients with an elevated B2M level have a shorter survival, and worse responses to traditional chemotherapy approaches. A summary of prognostic factors in CLL is provided in Table 28-4.
Immunophenotypic Features of Malignant Conditions Affecting Mature B Lymphocytes |
Disorder | Common immunophenotype |
CLL | DR+, CD19+, CD20+, CD5+, CD22−, CD23+, CD10−, weak sIg |
Prolymphocytic leukemia | DR+, CD19+, CD20+, CD5−, CD22+, CD23−, CD10−, bright sIg |
Mantle cell lymphoma | DR+, CD19+, CD20+, CD5+, CD22+, CD23−, CD10−, moderate sIg |
Follicular lymphoma | DR+, CD19+, CD20+, CD5−, CD22+, CD23−, CD10+, bright sIg |
Hairy cell leukemia | DR+, CD19+, CD20+, CD5−, CD22+, CD23−, CD10−, CD11c+, bright sIg |
CLL, chronic lymphocytic leukemia; sIg, surface immunoglobulin.
The mutational status of the IgVH locus is an important genetic parameter for prognostication in CLL patients, with the mutated IgVH (M-IgVH) associated with slow progression and long survival, whereas the un-mutated IgVH (UM-IgVH) is associated with an unfavorable course and rapid progression. Surrogate markers of UM-IgVH and poor prognosis include flow cytometry-detected expression of CD38 and ZAP-70 by CLL cells. Recent studies have suggested that more than 20% ZAP-70 expression as assessed by flow cytometry is associated with a median survival of less than 5 years, compared with 10 years in patients with less than 20% expression, and is now widely available. This was superior in predicting outcomes in CLL patients compared with IgVH mutational status testing.
Prognostic Factors in Chronic Lymphocytic Leukemia |
Favorable | Unfavorable |
Low Rai or Binet clinical stage | High Rai or Binet clinical stage |
Lymphocyte doubling time >12 mo | Lymphocyte doubling time <12 mo |
Nodular or interstitial BM infiltrate | Diffuse BM infiltrate |
Mutated IgVH | Unmutated IgVH |
ZAP-70 negative (low) | ZAP-70 positive (high) |
CD38 negative | CD38 positive |
13q− | 17p−/p53 abnormalities, 11q−/ATM abnormalities, +12 |
| Increased levels of: B2M, LDH, sCD23 Presence of NOTCH1, SF3B1, TP53, ATM mutations |
BM, bone marrow; ATM, ataxia telangiectasia mutated; LDH, lactose dehydrogenase; B2M, beta 2 microglobulin.
Interphase cytogenetics has identified both favorable (13q−) with a longer treatment-free interval and overall survival, and unfavorable (17p−, 11q−) with a short treatment-free interval and overall survival, subsets of CLL patients (Table 28-2). Consistent with this, subsets of patients with mutations in p53 (17p) or ATM (11q) have a poorer prognosis. When molecular parameters consisting of IgVH mutational status, 17p−, and 11q− were included in a multivariate analysis, the clinical stage was not identified as an independent prognostic factor. Four distinct molecular prognostic groups (17p−, 11q−, UM-IgVH, M-IgVH) have been identified, and have provided a framework for risk-adapted therapeutic strategies. Laboratory testing by interphase cytogenetics for genetic abnormalities of clinical significance in CLL patients is now widely available. Although these advances in genetic and molecular prognostic factors are promising, the decision to treat patients with CLL based upon them requires validation in prospective, randomized clinical trials. More recently, recurrent somatic mutations have been identified in CLL that have been integrated into cytogenetics-based risk scores. For example, a retrospective study of previously untreated CLL patients identified high risk (p53 or BIRC3 abnormalities), intermediate risk (NOTCH1 or SF3B1 mutations and/or 11q deletion), low risk (trisomy 12 or no cytogenetic abnormality), and very low risk (13q− as an isolated abnormality). These findings will require confirmation in prospective clinical trials, and assessment in relapsed/refractory patients.
E. Complications associated with chronic lymphocytic leukemia
1. Richter’s transformation. Richter’s syndrome (RS), originally described as the development of an aggressive NHL in patients with CLL/SLL, is now commonly used to describe transformation into any aggressive malignancy including diffuse large B-cell lymphoma (DLBCL), or, less commonly, PLL, Hodgkin’s lymphoma (HL), lymphoblastic lymphoma, hairy cell leukemia, or aggressive T-cell NHL. RS occurs in 2% to 8% of CLL patients, and results are conflicting on whether purine analog therapy increases the risk for transformation. In about 80% of patients, the malignant clone in RS develops through transformation of the original CLL clone (molecularly related to original CLL), while in about 20% of patients, they arise as an independent neoplasm. This distinction is clinically important, since patients with clonally unrelated RS/DLBLC have a prognosis similar to de novo
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


