Chimeric antigen receptor (CAR)-modified T cells targeting CD19 expressed by normal and malignant B cells is a unique therapy for patients with chronic lymphocytic leukemia (CLL); recent results highlight the potential of this therapy for patients with relapsed CLL. Because adoptive transfer of CAR-modified T cells is a novel approach, there are issues for the medical oncologist to consider when evaluating current and future clinical trials for CLL patients. This article reviews the impact of CAR design, T-cell production, T-cell dose, conditioning regimens, and tumor burden at the time of CAR-modified T-cell infusion on the efficacy of this therapy.
Key points
- •
Numerous targeted therapies are being developed for patients with chronic lymphocytic leukemia (CLL).
- •
CAR-modified T cells targeting CD19 expressed by normal and malignant B cells is a unique therapy, and recent results from 4 different trials highlight the dramatic potential of this therapy for patients with relapsed CLL.
- •
Because adoptive transfer of chimeric antigen receptor–modified T cells is a novel approach to cancer therapy, there are issues for the medical oncologist to consider when evaluating current and future clinical trials for patients with CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the target for numerous new investigational drugs and immunotherapies. Unique among these is the genetic modification of T cells to B-cell antigens through the gene transfer of a chimeric antigen receptor (CAR), which is composed of an antigen-binding component fused to T-cell signaling domains. A patient’s own T cells are genetically modified and then adoptively transferred back to the patient to mediate killing of malignant, and normal, B cells. Over the past 10 years, work initiated at the authors’ center has transitioned this technology from preclinical models to clinical trials, with evidence of promising results. However, there are important details that should be considered when evaluating and comparing the various CAR-modified T cells under study, because this therapy is unlike any traditionally used by the medical oncologist. The goal of this article is to describe and evaluate these details, including CAR design, T-cell production and dose, prior conditioning chemotherapy regimens, and tumor burden, and to discuss how they may affect the treatment response in patients with CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the target for numerous new investigational drugs and immunotherapies. Unique among these is the genetic modification of T cells to B-cell antigens through the gene transfer of a chimeric antigen receptor (CAR), which is composed of an antigen-binding component fused to T-cell signaling domains. A patient’s own T cells are genetically modified and then adoptively transferred back to the patient to mediate killing of malignant, and normal, B cells. Over the past 10 years, work initiated at the authors’ center has transitioned this technology from preclinical models to clinical trials, with evidence of promising results. However, there are important details that should be considered when evaluating and comparing the various CAR-modified T cells under study, because this therapy is unlike any traditionally used by the medical oncologist. The goal of this article is to describe and evaluate these details, including CAR design, T-cell production and dose, prior conditioning chemotherapy regimens, and tumor burden, and to discuss how they may affect the treatment response in patients with CLL.
Results of clinical trials
Clinical outcomes of 16 patients with CLL treated with CAR-modified T cells targeted to the B-cell–specific CD19 antigen have recently been reported from 4 trials conducted at various academic medical centers. The National Cancer Institute (NCI) reported their results concerning 4 patients with relapsed CLL treated with fludarabine and cyclophosphamide followed by CD19-targeted CAR-modified T cells. These patients, previously treated with an average of 4 chemotherapy regimens, had variable anti-CD19 responses including a complete remission (CR) of greater than 15 months’ duration. In addition, several patients developed anticipated B-cell aplasia as a consequence of their treatment and exhibited systemic serum cytokine elevations consistent with robust CAR-modified T-cell activation. Investigators at the University of Pennsylvania (UPenn) reported the results of 3 CLL patients treated with CD19-targeted CAR-modified T cells, of whom 2 patients had relapsed disease and 1 patient was chemotherapy-naïve, treated with bendamustine or pentostatin plus cyclophosphamide as conditioning therapy before T-cell infusion. Two of the patients had ongoing CR while the third achieved a partial remission (PR). Similar to the clinical outcomes at the NCI, 1 of these patients experienced a prolonged (>6 months) B-cell aplasia. The authors recently reported the largest cohort of CLL patients treated with CD19-targeted T cells ( Fig. 1 ). Outcomes in these patients included objective responses with lymph node reductions and B-cell aplasia. Furthermore, this trial included a unique secondary end point evaluating the requirement for conditioning therapy before gene-modified T-cell infusion. Lastly, investigators at the Baylor College of Medicine reported the results of 6 patients with B-cell malignancies, 1 of whom had CLL. Although no objective response was detected, the patient did have stable disease (SD) for 10 months after T-cell infusion. Of note, this trial did not include prior conditioning chemotherapy.
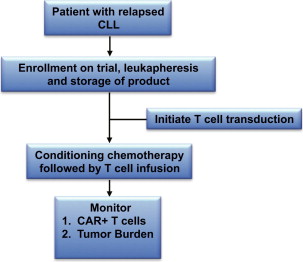
Overall, the toxicities reported among the different trials were quite similar and included fevers, rigors, hypotension, and B-cell aplasia. These toxicities began approximately 1 to 21 days after initial T-cell infusion. Furthermore, the toxicities appeared to be coincident with peaks in cytokine production. Collectively, in 2 of these patients their symptoms resolved and cytokine levels decreased after initiation of steroid therapy.
All of the reported trials present clinical evidence to support in vivo CD19-targeted T-cell efficacy. However, closer inspection of results and comparison of the trials focusing on elements of CAR design, T-cell production, and patient selection allows for a better understanding regarding disparities in results from the individual trials, providing insight for more rational designs of future CARs and therapeutic clinical trials. Furthermore, this discussion will allow the medical oncologist to critically evaluate the multiple clinical trials involving gene-modified T-cell therapy available for their patients with CLL ( Table 1 ).
| Clinical Trial Identifier | CAR | Gene Transfer | Disease Status | T-Cell Escalation | Conditioning Therapy | Trial Site |
|---|---|---|---|---|---|---|
| NCT00586391 | 19z a vs 1928z | γ-Retrovirus | Relapsed | Yes | CY | Dallas, TX |
| NCT00709033 | 19z b vs 1928z | γ-Retrovirus | Relapsed | Yes | CY | Dallas, TX |
| NCT00924326 | 1928z | γ-Retrovirus | Relapsed | No | CY + FLU Interleukin-2 c | Washington, DC |
| NCT00968760 d | 1928z | Electroporation with SB transposase | Relapsed | Yes | BEAM + R ± Interleukin-2 | Houston, TX |
| NCT01653717 | 1928z | Electroporation with SB transposase | 8 weeks from last chemo | Yes | CY + FLU | Houston, TX |
| NCT01416974 | 1928z | γ-Retrovirus | MRD e | Yes | CY | New York, NY |
| NCT00466531 | 1928z | γ-Retrovirus | Relapsed | Yes | CY | New York, NY |
| NCT01029366 | 19BBz | Lentivirus | Relapsed | No | Investigators’ choice | Philadelphia, PA |
a Patients are infused with a mixture of T cells modified with either the 19z or 1928z CAR.
b Patients are infused with a mixture of T cells modified with either the 19z or 1928z CAR. The 19z CARs are transduced into EBV+ T cells, whereas the 1928z CARs are transduced into normal peripheral, polyclonal T cells.
c Interleukin-2 is not given as a lymphodepleting agent but as a T-cell growth factor.
d This is the only trial listed in which the T cells are administered as part of an autologous stem cell transplant.
e This trial is evaluating CAR-modified T cells as a consolidation regimen. Patients treated with an initial chemotherapy regimen and who have residual disease after completing this regimen are infused with 1928z+ T cells.
CAR design
CARs are generally classified as being of first-generation, second-generation, or third-generation design. This classification relates to the signal transduction domains incorporated within the CAR ( Fig. 2 ). First-generation CARs most commonly consist of a CD3ζ signaling element, which when combined with an anti-CD19 single chain variable fragment (scFv) successfully redirects T cells to mediate killing of B cells in vitro and in vivo in immunodeficient preclinical animal models. However, these first-generation CARs ultimately have been found to have limited in vivo efficacy, with little evidence of T-cell persistence in these models. The reason for this limited efficacy is related to T-cell biology: T cells are optimally activated when they encounter antigen for the first time if they receive two signals, one mediated by CD3ζ (signal 1) and the other mediated by a costimulatory receptor, most commonly CD28 (signal 2). This 2-signal paradigm for efficient T-cell activation could be recapitulated through second-generation CARs that included costimulatory T-cell cytoplasmic signal domains proximal to CD3ζ cytoplasmic signal domains (see Fig. 2 ). T cells modified to express second-generation CARs demonstrated enhanced in vivo tumor killing and persistence. While CD28 is the most commonly used costimulatory signaling domain, others have modified second-generation CARs to include the costimulatory signal domains of 41BB, OX40, DAP10, and CD27. Studies have demonstrated that additional signal domains enhance gene-modified T-cell function by increasing cytokine secretion and enhancing T-cell proliferation and persistence. Third-generation anti-CD19 CARs, which have 2 costimulatory domains combined with CD3ζ, demonstrate impressive results in preclinical animal models, but have not been evaluated in CLL patients to date.
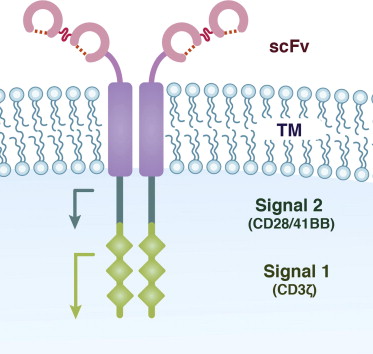
Comparison of anti-CD19 CARs using different monoclonal antibody (mAb) derived scFvs have not been performed, although one could speculate that if the binding affinities of the scFvs were significantly different it could affect CAR-mediated T-cell activation and consequent B-cell killing. To this end, studies at the Memorial Sloan-Kettering Cancer Center (MSKCC) used a different scFv, derived from the SJ25C1 hybridoma, in comparison with studies at the NCI and UPenn wherein the anti-CD19 CAR used a scFv derived from the FMC63 hybridoma.
The 4 clinical trials involving CLL patients have all used second-generation CARs, but the clinical trial results reported by Savoldo and colleagues are unique for directly infusing a mixture of T cells genetically modified with a first-generation CD3ζ CAR and a second-generation CAR including the CD28 costimulatory domain. In a cohort of 46 patients (1 with CLL), investigators clearly demonstrated that T cells with second-generation CARs enhanced persistence and/or expansion when compared with T cells modified with a first-generation CAR.
Investigators at UPenn have the only trial for CLL patients using a CAR that has a costimulatory domain other than CD28, namely 41BB. At present the only direct comparison of anti-CD19 second-generation CARs with a CD28 or 41BB costimulatory domain (19-28z vs 19-bbz) is in preclinical models, and the results documenting protection against B-cell malignancies have been contradictory, possibly because the anti-CD19 scFvs were derived from different mAbs.
T-cell production
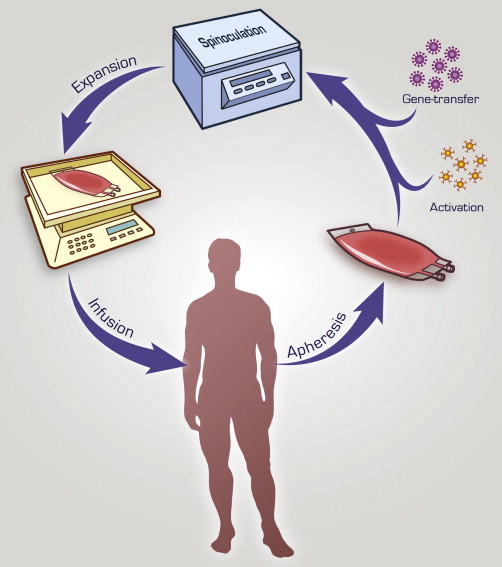
In most trials, CAR-modified T cells are generated ex vivo and include an initial activation step followed by a gene-transfer step ( Fig. 3 ). All trials activate T cells with agonistic mAb-mediated CD3 stimulation with or without additional CD28 costimulation. In 3 of the reported clinical trials gammaretroviral vectors were used for gene transfer, whereas studies from UPenn used lentiviral vectors. However, given the small number of patients treated to date on these trials, it is not yet possible to assess the superiority of one viral transfer system over the other. Although in theory lentiviral gene transfer may increase safety given prior reports of leukemogenic integration sites associated with gammaretroviruses, in these cases the cells transduced were hematopoietic stem cells, not mature T cells. To date, there have been no reports of insertional oncogenesis with gammaretroviral vectors in the context of genetically modified mature lymphocytes. In fact, a recent report identified no long-term sequelae in 43 subjects infused with gammaretroviral transduced T cells in several clinical trials evaluating patients after an 11-year follow-up period.
Related posts:
 The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
 The Role of Minimal Residual Disease Measurements in the Therapy for CLL
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Chimeric Antigen Receptor Therapy for Chronic Lymphocytic Leukemia
The Role of Minimal Residual Disease Measurements in the Therapy for CLL
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Chimeric Antigen Receptor Therapy for Chronic Lymphocytic Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


