96 Dario Campana and Ching-Hon Pui • Leukemia is the most common childhood cancer. • The most common subtype, acute lymphoblastic leukemia (ALL), accounts for 75% to 80% of all cases of childhood leukemia, whereas acute myeloid leukemia (AML) comprises approximately 20%. • Chronic myelogenous leukemia (CML), myelodysplastic syndrome (MDS), and myeloproliferative disorder (MPD) are less frequent. • Chronic lymphocytic leukemia (CLL) is extremely rare. • Although environmental agents, such as ionizing radiation and chemical mutagens, have been implicated in the induction of leukemia, discernible etiologic factors are lacking in almost all cases of primary leukemia. • Acquired genetic changes are central to the development of leukemia. • Males are generally affected by leukemia slightly more often than females in all age groups except infants. • In developed countries, the incidence of ALL is highest between ages 2 and 5 years. • The incidence of AML is relatively constant during childhood, with slight peaks in the first 2 years of life and in late adolescence. • MDS typically occurs after 5 years of age and juvenile myelomonocytic leukemia (JMML) occurs nearly always before 5 year of age. • Physical signs and symptoms of thrombocytopenia and anemia are common. • Neutropenia may lead to severe infection. • Bone pain and arthralgia caused by leukemic infiltration is more common in ALL than AML and may be especially severe in young children. • Common sites of extramedullary involvement in ALL include liver, spleen, thymus, and lymph nodes. • Skin, gums, and the head and neck area are typical sites of extramedullary disease in AML Infiltration of the central nervous system can be found in both ALL and AML. • The acute onset of petechiae, ecchymoses, and bleeding may suggest idiopathic thrombocytopenic purpura. • Both acute leukemia and aplastic anemia may present with pancytopenia and complications associated with bone marrow failure. • Infectious mononucleosis and other viral infections can be confused with ALL. • Bone pain, arthralgia, and occasionally arthritis may mimic juvenile rheumatoid arthritis, rheumatic fever, other collagen diseases, or osteomyelitis. • Childhood ALL should also be distinguished from pediatric small round cell tumors that involve the bone marrow. • Patients with ALL undergo a relatively brief remission-induction phase followed by intensification (consolidation) therapy and then prolonged continuation treatment. • All patients require treatment for subclinical central nervous system (CNS) involvement, which should be initiated early in the form of intrathecal chemotherapy. • Most protocols for AML include remission induction and 3 to 4 courses of consolidation therapy. • Autologous hematopoietic stem cell transplantation is not usually recommended. • At present, ALL with remission failure, high levels (≥1%) of minimal residual disease (MRD) and/or persistent MRD postremission induction or early hematologic relapse are clear indications for allogeneic transplantation. • Allogeneic transplantation appears to improve overall survival in AML, although the indications for this procedure during first remission are debated. • Five-year event-free survival estimates for children with newly diagnosed ALL are now over 80%. • Hypodiploidy with less than 44 chromosomes, and early T-cell precursor status are unfavorable prognostic indicators, whereas hyperdiploidy with more than 50 chromosomes and the ETV6-RUNX1 gene fusion in B-lineage ALL and NOTCH/FBXW7 mutations in T-cell ALL are associated with a favorable outcome. The prognosis associated with the Philadelphia chromosome/BCR-ABL1 abnormality has improved with the availability of tyrosine kinase inhibitors. • Event-free survival for infant ALL with 11q23/MLL rearrangement remains only 20% to 35% and has not been improved by allogeneic transplantation. • In AML, patients with Down syndrome or acute promyelocytic leukemia have a favorable prognosis with optimal therapy, whereas those with acute megakaryoblastic leukemia without the t(1;22)/OTT-MAL have significantly worse outcomes than others. • Early bone marrow relapse and treatment-related AML carry a dismal prognosis. • Patients with MDS, AML arising from MDS, AML with monosomy 7, or AML with internal tandem duplication of the FLT3 gene often have resistant disease. • Slow response to remission induction therapy and persistent minimal residual disease are associated with a higher risk of relapse in both ALL and AML. Acute leukemia is a malignant proliferation and accumulation of immature lymphohematopoietic cells. The leukemic cell population is shown to be clonal by cytogenetics, glucose-6-phosphate dehydrogenase characterization, and analysis of antigen-receptor gene rearrangements and X-linked restriction fragment-length polymorphisms.1 Although leukemic cells generally do not proliferate as actively as their normal hematopoietic counterparts,2 they accumulate inexorably and compete successfully with normal cells. Their inability to differentiate and their relative resistance to apoptosis may explain this phenomenon. By the time of diagnosis, leukemic cells have usually replaced normal bone marrow cells and disseminated to various extramedullary sites. Therefore the presenting features of leukemia typically reflect the degree of bone marrow replacement and the extent of extramedullary spread. Both ALL and AML are heterogeneous diseases that comprise different biological subtypes. The major morphologic and immunophenotypic divisions based on lineage association and degree of maturation are subclassified by the identification of distinct, recurrent chromosomal and molecular abnormalities,3,4 and gene expression patterns.5–9 Chronic myelogenous leukemia (CML), myelodysplastic syndrome (MDS), and myeloproliferative disorders (MPD) are infrequent in children. In general, they share clinical, laboratory, and morphologic features with one of their adult counterparts.10–13 A disorder characterized by an heterogeneous pattern of myeloproliferation, dysplasia, and hepatosplenomegaly in association with abnormal peripheral blood counts has historically been called “juvenile CML,” “infantile monosomy 7 syndrome,” and “chronic myelomonocytic leukemia” by different authors; the term juvenile myelomonocytic leukemia (JMML) is currently favored.14 Only a few well-documented cases of chronic lymphocytic leukemia have been reported in children.15,16 Leukemia is the most common malignancy among patients younger than 15 years. ALL is approximately four times more common than AML. Males are generally affected by leukemia slightly more often than females in all age groups, with two exceptions: boys have a risk of T-cell leukemia that is four times that of girls, and girls have a slightly higher incidence of leukemia in the first year of life (1.5 : 1 ratio).17 Rates of ALL are comparatively higher in Northern and Western Europe, North America, and Oceania, than in Asia and Africa.17 In developed countries, the incidence of ALL is highest between ages 2 and 5 years. This age peak is accounted for largely by ALL with hyperdiploidy (>50 chromosomes) or ETV6-RUNX1 (formerly known as TEL-AML1) gene fusion.3 The incidence of ALL is higher in the Hispanic than in the white population, which, in turn, is higher than in the black population, especially among children 2 to 5 years of age. Black children have a higher incidence of T-cell ALL and pre-B leukemia with E2A-PBX1 fusion, and are less likely to have hyperdiploid ALL with more than 50 chromosomes than are white children.18 The incidence of AML in children peaks at 2 years of age, decreases to a nadir at 9 years, then peaks again at around age 16 years.19 The highest incidence of pediatric AML occurs among the Maori of New Zealand, Hawaiian Americans, and Africans in Zimbabwe.19 In most populations of children, less than 10% of cases of AML are acute promyelocytic leukemia.20 However, this figure is approximately 25% in children of Latin American origin21; a higher incidence was also reported in Italian children.22 Pediatric patients with MDS are typically older at diagnosis (>5 years old), whereas JMML nearly always occurs before 5 years of age.14 A small percentage (<5%) of cases of leukemia are associated with inherited genetic syndromes (Table 96-1). Children with Down syndrome have a tenfold to twentyfold increased risk of leukemia (ALL and AML).23,24 Furthermore, a MPD termed transient abnormal myelopoiesis may occur in newborns with Down syndrome.25 This disorder spontaneously remits in almost all patients within 3 months. However, early death can occur in almost 20% of patients with high presenting leukocyte count, abnormal liver function, and failure to normalize blood count; AML subsequently develops in another 30% of patients.25 Several other genetic disorders are associated with an increased risk of leukemia and/or MDS, including ataxia-telangiectasia, Wiskott-Aldrich syndrome, Bloom syndrome, Fanconi anemia, Kostmann disease, Blackfan-Diamond anemia, and neurofibromatosis.19,26,27 The association between leukemia and congenital immunodeficiencies, such as X-linked agammaglobulinemia and common variable immunodeficiency, is not well supported.26 Table 96-1 Congenital Disorders Associated with Increased Risk of Leukemia Fraternal twins and siblings of affected children are at a twofold to fourfold greater risk of leukemia during the first decade of life than unrelated children.17,28 However, a Nordic population- and register-based study failed to show an increased incidence among siblings of children with leukemia.29 When leukemia occurs in one identical twin, the likelihood that the other twin will develop the disease is approximately 20%. However, when leukemia is diagnosed in one twin before 1 year of age, it almost invariably develops in the other twin, typically within a few months. Molecular studies have demonstrated that intrauterine metastasis of ALL from one twin to the other, via the shared placental circulation, is responsible for the concordant leukemia.32–32 Although environmental agents, such as ionizing radiation and chemical mutagens, have been implicated in the induction of leukemia, discernible etiologic factors are lacking in almost all cases of primary leukemia.17 Association between leukemia and maternal exposure to various potential mutagens, neonatal administration of vitamin K, parental use of medications and drugs, and proximity to electromagnetic fields has not been authoritatively demonstrated.17 Although an association between increased risk of childhood ALL and exposure to high levels of residential magnetic fields has been reported, this finding needs confirmation.35–35 A case-control study identified a significantly increased risk of leukemia among the offspring of men employed in occupations with a risk of exposure to electromagnetic fields or radiation,36 a finding that also warrants further studies. Because industrialization, higher socioeconomic status and social isolation are associated with an increased risk of B-lineage ALL, Greaves hypothesized that abnormally late exposure to common infections and immune response stimulation could enhance the likelihood of leukemogenic genetic mutations, thus increasing the risk of childhood B-lineage ALL.37 In line with this notion is the observation of occasional clustering of childhood ALL associated with rural-urban population mixing, especially in new towns.38,39 Infant leukemias frequently have rearrangements of the MLL gene, located on chromosome band 11q23.40,41 MLL rearrangements are also common in therapy-related AML, arising after treatment with topoisomerase II inhibitors.42 This molecular similarity raised the hypothesis that transplacental fetal exposure to substances that inhibit topoisomerase II, such as flavonoids (in food and drink), quinolone antibiotics, benzene metabolites, catechins, and estrogens, could be leukemogenic.41 A case-control study found that in utero exposure to DNA-damaging drugs, herbal medicines, or pesticides was significantly associated with infant leukemia with MLL rearrangements.43 Because the functional doses received via dietary and environmental exposure are much lower than those received from anticancer chemotherapy, it was postulated that affected infants or their mothers may have reduced activity of carcinogen-detoxifying enzymes because of genetic polymorphism. In this regard, deficiency of glutathione-S-transferases (GST-M1 and GST-T1), enzymes that detoxify electrophilic metabolites by catalyzing their conjugation to glutathione, is associated with infant leukemia without MLL rearrangement,41 and with ALL in black children.44 Polymorphisms of reduced nicotinamide adenine dinucleotide phosphate : quinone oxidoreductase, an enzyme that converts benzoquinones to less toxic hydroxyl metabolites, have been associated with the development of infant and childhood ALL.45,46 Cytochrome P-450 CYP1A1*2A and NQO1*2 variant genotypes have also been linked to an increased risk of childhood ALL; children carrying both genotypes were at a particularly high risk.47 It has also been suggested that folate pathways may play a role in susceptibility to ALL,48,49 and that folate supplement may reduce the risk,50 an intriguing finding that requires confirmation. However, recent genome-wide associated studies could not confirm those findings and instead identified a number of other inherited polymorphisms in genes such as ARID5B, IKZF1, CEBPE, and CDKN2A, that are associated with the risk of childhood ALL in various ethnic and racial groups.51–54 To date, no direct gene-environment interaction has been firmly established. There is strong evidence that acquired genetic changes are central to the development of leukemia. These changes affect the number (ploidy) and/or the structure of chromosomes; structural changes comprise translocations, inversions, deletions, point mutations, and amplifications.55 The dysregulation of genes encoding transcription factors and the resulting subversion of transcriptional pathways that regulate hematopoietic cell homeostasis, provides a mechanistic explanation for leukemogenesis.56,57 For example, the core binding factor (CBF) family of transcription factors is disrupted by recurrent chromosomal translocations, such as those involving ETV6-RUNX1 in ALL and RUNX1-RUNX1T1 and CBFB–MYH11 in AML.57,58 The encoded proteins regulate the expression of growth factors, such as interleukin-3, granulocyte-macrophage colony-stimulating factor, and macrophage colony-stimulating factor receptor, as well as the T-cell receptor β enhancer and the immunoglobulin heavy chain enhancer/promoter. The function of homeobox (HOX) genes, an evolutionarily highly conserved family of transcription factors whose expression is tightly regulated during hematopoietic cell differentiation, can also be disrupted either by direct involvement in chromosomal translocations [e.g., the t(7;11), forming the NUP98-HOXA9 fusion gene] or by the disruption of proteins believed to be their upstream regulators.59 Among the latter, the most notable is encoded by MLL, a gene crucial for both embryonic development and hematopoiesis, and, as mentioned above, is involved in translocations of the 11q23 region in both ALL and AML.58,59 Chimeric proteins encoded by fused genes can also directly interfere with normal apoptotic pathways, as has been demonstrated in the case of the TCF3-HLF protein.60,61 The expression of key regulators can be altered in the absence of detectable genetic abnormalities. For example, five different T-cell oncogenes (HOX11, TAL1, LYL1, LMO1, and LMO2) can be aberrantly expressed in T-lineage ALL in the absence of chromosomal abnormalities.6 Cell survival requirements can be altered by the dysregulated activity of tyrosine kinases, as in the case of the BCR-ABL1 gene fusion.4 Alternatively, activating mutations in tyrosine kinase receptors for growth factors may confer a growth advantage to leukemic cells. This mechanism is exemplified by mutations of FLT3, which encodes a receptor tyrosine kinase expressed by immature hematopoietic cells that acts synergistically with other growth factors to stimulate proliferation of hematopoietic progenitor cells.62 Mutant FLT3 is detectable in both ALL and AML; these mutations typically involve small tandem duplications of amino acids that result in constitutive tyrosine kinase activity.62 Activating mutations of NOTCH1, a gene encoding a transmembrane receptor that regulates normal T-cell development, are frequently detected in T-cell ALL.63,64 Activating mutations of NOTCH1 produce constitutive NOTCH1 signaling, which is sufficient to induce T-cell ALL in experimental models.64 Gamma secretase, a multicomponent membrane-associated enzyme, is required for NOTCH1 signaling through mutant NOTCH receptors in T-cell ALL, providing a target for therapeutic intervention with γ-secretase inhibitors.65 NOTCH signaling can also be interfered with by directly targeting NOTCH receptors with specific antibodies,66 thus avoiding the toxicity associated with γ-secretase inhibitors. Interestingly, somatic mutations inactivating the NOTCH pathway have been identified in some patients with chronic myelomonocytic leukemia, suggesting that NOTCH can also have tumor-suppressive functions.67 A pioneering large-scale study of DNA single-nucleotide polymorphism (SNP) analysis examined 242 cases of pediatric ALL and identified lesions in genes encoding key regulators of B-cell differentiation in 40% of B-cell precursor ALL cases.68 Most prominent were deletions and cryptic translocations involving the PAX5 gene, which was altered in almost one third of cases, and which would be predicted to block normal B-progenitor cell differentiation before immunoglobulin heavy-chain gene rearrangement. Other mutated genes were found in concert with several of the more common translocation-induced chimeric oncogenes, such as TCF3-PBX1 and ETV6-RUNX1, including the essential B-cell developmental genes E2A, EBF, LEF1, Ikaros, and Aiolos. The retrospective identification of leukemia-specific fusion genes (e.g., MLL-AFF1, ETV6-RUNX1) in the neonatal blood spots of identical twins who experienced concordant leukemia has demonstrated their prenatal origin.32,69 In cases with the t(4;11) and MLL-AFF1, there is a high rate of concordance in identical twins (25% to 100%) and a very brief latency period after birth (a few weeks to a few months).32 In other types of leukemia, for example, those with the ETV6-RUNX1 fusion or T-cell phenotype, the rate of concordance is lower, the postnatal latency period is longer and is variable, and the clinical presentation and the outcome of therapy may differ widely among identical twins, suggesting that secondary postnatal molecular events are necessary for full leukemic transformation.32 Further insights were gained from a report of a set of triplets in which the two monozygotic twins developed concordant leukemia with identical ETV6-RUNX1 fusion at 3 years of age, while the third child, who had developed from a second zygote, was free of leukemia and of the genomic sequence.31 In addition to the fusion transcript, the identical twins had a secondary, independent deletion of the normal unrearranged ETV6 allele, suggesting a contributing postnatal event. Clone-specific antigen receptor gene rearrangements were analyzed in the neonatal blood spots of five children who were 6 months to 4 years and 8 months of age at diagnosis of B-lineage ALL and T-ALL: in all five children, the clonotypic antigen receptor gene rearrangements had been present at birth and the estimated number of clonotypic cells per blood spot was in the range of 10 to 100.70 One study found the ETV6-RUNX1 fusion in the cord blood of approximately 1% of randomly selected newborns, a frequency approximately 100 times that of ALL with ETV6-RUNX1,71 further supporting the notion that preleukemic clones are generated in utero at a high frequency and that secondary postnatal leukemogenic events are required for a fully malignant transformation of ETV6-RUNX1 cells. The prevalence of leukemias with prenatal origin is not known and not all cases develop in utero. For example, t(1;19) E2A-PBX1 ALL appears to have a postnatal origin in most cases.72 There is also evidence for RUNX1-RUNXT1 gene fusions occurring in utero,73 but the precise frequency of in utero events for this and other type of genetic abnormalities associated with AML is unclear.74 Leukemic blast cells are identified at diagnosis in the cerebrospinal fluid (CSF) of as many as one third of children with ALL (most of whom have no neurologic symptoms) and in approximately 5% of children with AML. Traditionally, CNS leukemia is defined by the presence of at least 5 leukocytes per microliter of CSF and the detection of leukemic blast cells, or by the presence of cranial nerve palsy. However, the presence of any amount of leukemic cells in CSF, even from iatrogenic introduction as the result of a traumatic lumbar puncture, is associated with an increased risk of ALL relapse, and requires additional intrathecal therapy.75,76 Pancytopenia is a presenting feature in most cases of MDS.13 Fetal hemoglobin is frequently slightly elevated. Most patients have no organomegaly. Extramedullary myeloid tumor may be the presenting feature of MDS but blasts in the CSF is not seen in MDS. The bone marrow is usually normocellular or hypercellular, with characteristic dysplastic features, including megaloblastic erythropoiesis, bizarre small or unusual large megakaryocytes, and dysgranulopoiesis; the percentage of myeloblasts is often increased.13 Hepatosplenomegaly and lymphadenopathy are suggestive clinical features of JMML.14 Current World Health Organization diagnostic criteria for patients with JMML include a peripheral monocyte count greater than 1 × 109/L with less than 20% bone marrow blasts and splenomegaly in the absence of BCR-ABL1. Other characteristic features of JMML are somatic mutation in RAS or PTPN11, clinical diagnosis of NF1 or NF1 gene mutation, and/or monosomy 7. A feature of JMML cells is their capacity to spontaneously form granulocyte-macrophage colonies in vitro, and their hypersensitivity to granulocyte-macrophage colony-stimulating factor.14 MDS with a low blast count must be distinguished from aplastic anemia and other nonclonal disorders, which often requires sequential morphologic studies, including bone marrow biopsies.13 Myelodysplasia may also occur in a variety of disorders, such as infection, drug therapy, and chronic disease. AML is the major differential diagnosis of MDS. Cases with AML-specific translocations are more appropriately classified as AML regardless of the blast count, whereas monosomy 7 as the only cytogenetic aberration is strongly suggestive of MDS.13 A definitive distinction between JMML and Philadelphia chromosome-positive CML requires karyotypic or molecular examination for the t(9;22)/BCR-ABL1 abnormality. These conditions also must be distinguished from the “leukemoid reaction” associated with infection, cancer, or congenital heart disease in which signs and symptoms do not progress and/or resolve spontaneously.14 Morphologic analysis of leukemic cells in smears stained with Romanowsky stains (Wright-Giemsa or May-Grünwald-Giemsa) distinguishes three subtypes of ALL (L1, L2, and L3) and eight subtypes of AML (M0-M7) as classified by the French–American–British (FAB) scheme (Fig. 96-1).77 However, the FAB classification for ALL is no longer used because the defining criteria are too subjective, and it has no prognostic or therapeutic significance. The term acute myeloid leukemia is used to designate even leukemias in which some or all cells have the morphology of monocytes (M4, M5), erythroblasts (M6), or megakaryoblasts (M7). Because of the rarity and heterogeneous nature of childhood MDS, its classification has been inconsistent.13 Analysis of a Romanowsky-stained smear cannot accurately distinguish between ALL and AML. Cytochemical stains help this distinction. Myeloperoxidase, Sudan black, and nonspecific esterases, including α- naphthyl butyrate and α-naphthyl acetate esterase, react with myeloid blast cells, whereas periodic acid-Schiff reagent reacts positively in more than 70% of ALL cases. However, despite the traditional use of morphology and cytochemistry, contemporary classification of acute leukemias is based on subtypes that can be identified only by immunologic and molecular analyses. The FAB group classified MDS into five subgroups: refractory anemia (RA), RA with ringed sideroblasts (RARS), RA with excess of blasts (RAEB), RAEB in transformation (RAEB-T), and chronic myelomonocytic leukemia.78 AML was defined by the presence of 30% myeloblasts in the bone marrow. A more recent World Health Organization classification classified myelodysplastic and myeloproliferative disorders into MDS, JMML, and myeloid leukemias of Down syndrome (Table 96-2).10,13 The distinction between MDS and AML can be difficult and may require molecular genetic studies. The presence of greater than 30% of blasts in the blood or marrow suggests AML, whereas less than 20% blasts suggests MDS.13 Table 96-2 Classification of Pediatric Myelodysplastic and Myeloproliferative Disease Proposed by Hasle et al. Data from Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003;17:277–82. Table 96-3 summarizes antigen expression patterns in ALL. Table 96-3 Immunophenotypic Subgroups of ALL *Detectable on the cell surface membrane in some cases. Data from Campana D, Behm FG. Immunophenotyping of leukemia. J Immunol Methods 2000;243:59–75. Leukemic blast cells of early pre-B ALL resemble normal marrow B-cell precursors. Immunoglobulin heavy-chain genes are usually rearranged in these cells but immunoglobulins are not detectable. The leukemic cells of early pre-B ALL always express CD19. Almost all cases have cytoplasmic CD22 and CD79α; surface CD22 expression is also evident in many cases.79 CD10 and terminal deoxynucleotidyl transferase (TdT) are detectable in 90% of cases, and cells in more than 75% of cases express CD34.79 The CD20 antigen is present on a minor proportion of blast cells in one-half of cases.79,80 In 10% to 15% of early pre-B ALL cases, CD45 is very weakly expressed or undetectable; cells that have this immunophenotype are usually hyperdiploid (modal chromosome number >50).79 ALL with rearrangement of the MLL gene typically has an early pre-B ALL phenotype with distinctive features such as expression of CD15, CD65 and surface chondroitin proteoglycan sulfate, and absence of CD10.79 Approximately 25% of newly diagnosed cases of ALL have a pre-B immunophenotype consisting of accumulation of cytoplasmic immunoglobulin µ heavy chains with no detectable surface immunoglobulins.79 Similar to early pre-B ALL, cells express CD19, CD22, and CD79α. Rearrangement of immunoglobulin light chain genes is evident in some of these leukemias, but κ and λ proteins are not detectable. More than 95% of pre-B ALL express CD10 and TdT, but only two-thirds express CD34.79 In many cases of pre-B ALL, surface CD20 is absent or is weakly expressed.79,80 ZAP-70 expression can be found in cases of B-lineage ALL, particularly those with a pre-B phenotype.81,82 Between 20% and 25% of pre-B ALL cases have either the t(1;19)(q23;p13) or the der(19)t(1;19)(q23;p13).79 The antigen expression profile CD19+, CD22+, CD20±, CD34−, CD45+, cytoplasmic µ+ is characteristic of ALL cases with the t(1;19), but is not specific to these cases.79 Leukemic cells that express both cytoplasmic and surface immunoglobulin µ heavy chains without κ or λ light chains have been designated transitional pre-B ALL,79 but this nomenclature is not widely used and there is no identified genetic abnormality that is characteristic of this subgroup of ALL. In 2% to 4% of childhood ALL cases, cells express surface immunoglobulin µ heavy chains plus either κ or λ light chains. The most common type of B-cell (Burkitt) ALL is characterized by L3 morphology according to the FAB classification. Cells express CD19, CD22, CD20, and frequently CD10 and CD23; CD34 is negative. In rare cases, TdT is expressed, or surface immunoglobulin is absent.79 B-cell ALL often represents the leukemic phase of Burkitt lymphoma arising in the abdomen or the head and neck.79 The hallmark of this subset of B-cell ALL is the presence of a reciprocal translocation of chromosome 8 with one of the chromosomes containing an immunoglobulin gene. These translocations, which include the t(8;14)(q24;q32), t(2;8)(p12;q24), and t(8;22)(q24;q11), involve rearrangement of the c-MYC gene. The less common subtype of B-cell ALL is characterized by blast cells with L1 or L2 morphology. These leukemias may express TdT and CD34, and they express CD20 only weakly.79 Extramedullary masses are not seen at presentation. The t(8;14), t(2;8), and t(8;22) are absent, as are characteristic rearrangements of the c-MYC gene. These cases should be treated as B-cell precursor ALL rather than Burkitt ALL. T-lineage ALL cells have surface CD7 and cytoplasmic CD3 (cCD3) antigens.83,84 More than 90% of T lymphoblasts express CD2, CD5, and TdT. Surface CD1a, CD3, CD4, and CD8 are detected in fewer than 45% of cases.79 The HLA-DR antigen is not commonly expressed, and 40% to 45% of cases are CD10+ and/or CD21+.79 CD79α is also expressed in approximately one-third of cases.79 T-lineage ALL has been divided into three stages of immunophenotypic differentiation: early (CD7+, cCD3+, surface CD3−, CD4−, and CD8−), mid or common (cCD3+, surface CD3−,CD4+, CD8+, and CD1+), and late (surface CD3+, CD1−, and either CD4+ or CD8+). However, as many as 25% of cases of T-lineage ALL have antigen patterns that do not conform to any of these maturation stages. T-cell receptor (TCR) proteins are heterogeneously expressed in T-lineage ALL.85 In approximately two-thirds of cases, membrane CD3 and TCR proteins are absent. In half of these cases, however, TCR proteins (TCRβ, TCRα, or both) are present in the cells’ cytoplasm. Most cases with membrane CD3 and TCR chains express the αβ form of the TCR, whereas a minority express TCRγδ proteins. Several early studies have yielded conflicting conclusions about the prognostic significance of the expression of surface CD3 and the absence of CD2, CD5, or CD10.88–88 More recently, a new subtype of T-ALL was identified, named early T-cell precursor (ETP)-ALL. ETP-ALL lymphoblasts express CD3 (typically cytoplasmic ) and lack myeloperoxidase.89 The defining features of ETP-ALL are absent expression of CD1a and CD8, dim CD5 expression (i.e., mean fluorescence intensity at least 10-fold lower than that of normal T lymphocytes or expression in less than 75% of blasts), and expression of stem cell/myeloid-associated markers, such as CD34, CD133, HLA-DR, CD13, CD33, and CD11b.89 ETP-ALL represents approximately 12% of T-ALL cases.89 Patients with this leukemia subtype have a dismal outcome, with poor response to initial therapy and a very high risk of relapse.89 Table 96-4 summarizes the patterns of antigen expression in AML. The leukemic cells in all myelocytic and monocytic subtypes of AML (M0 through M5) express various combinations of CD13, CD33, CD65, CD117, and myeloperoxidase (MPO).79 Table 96-4 Immunophenotype of AML Subtypes Data from Campana D, Behm FG. Immunophenotyping of leukemia. J Immunol Methods 2000;243:59–75. Approximately 35% to 45% of cases of childhood M2 AML have the t(8;21)(q22;q22). Leukemic blast cells commonly express MPO, CD34, CD65, and HLA-DR, but CD13 and CD33 expression is characteristically weak and sometimes is not detectable.79 Most cases weakly express CD19 and, less commonly, CD56.79 By contrast, the leukemic myeloblasts of M2 AML without the t(8;21) may also express MPO, CD34, CD65, and HLA-DR, but the expression of CD13 and CD33 usually exceeds that of myeloblasts that have the t(8;21). In addition, the CD19 antigen is rarely detectable, whereas T-cell−associated CD2 or CD7 is commonly present in these cases.79 This group of leukemias includes a microgranular variant referred to as M3v that may morphologically mimic acute monocytic leukemia. Cells of M3 and M3v AML strongly express MPO, CD13, CD33, and CD65 but usually lack HLA-DR.79,90 Expression of CD11b and CD15 is variable, and CD4 and CD56 are seldom detected.79 Atypical expression of CD2 is observed in 40% to 45% of cases, but may be more prevalent in the M3v subtype.79 CD34 is generally absent but may be found in some cases, usually M3v.90 Heterogeneous expression of CD13, the existence of a single primary blast cell population, and a characteristic pattern of CD34 and CD15 expression are reportedly useful in identifying M3 AML.91 Blast cells of most myelomonocytic leukemias express MPO, CD4, CD11b, CD11c, CD13, CD14, CD33, CD34, CD45, CD65, and HLA-DR.79 A relatively uncommon variant of M4 AML, M4Eo, is associated with increased numbers of eosinophils in the bone marrow, with or without peripheral blood eosinophilia. These cases usually express the CBFB-MYH11 chimeric gene, which is often associated with expression of CD2.79 Monoblasts usually express MPO, HLA-DR, CD4, CD11b, CD11c, CD33, and CD65. The cells of some monocytic leukemias express CD117, but CD34 is rarely detected. The cells of most monocytic leukemias also express CD15, CD36, and, not infrequently, CD56. Expression of CD14 is largely restricted to cells of the monocytic lineage, but is often absent in pediatric M5 cases. A variable number of monoblasts may appear to react with antibodies to CD41a and CD61 because of platelet adhesion to the cell surface or glycoprotein IIb/IIIa absorption.79 Leukemias composed primarily of erythroid precursors are uncommon. Leukemic erythroblasts usually express CD36, CD71, and glycophorin A, and hemoglobin is detectable in late-stage erythroid precursors. Cells of the myeloid component express CD13, CD33, and MPO. M6 AML may be difficult to distinguish from M0 and M7 AML, because undifferentiated erythroblasts have few or no erythroid-associated antigens, and their antigenic and ultrastructural features may mimic those of early megakaryoblasts. 79 Distinguishing acute megakaryoblastic leukemia (M7 AML) from ALL, M0 and M5 AML, acute erythroblastic leukemia and metastatic small-cell tumors solely on the basis of cell morphology might be difficult. The leukemic cells of most M7 AML cases express CD41a and CD61, and those of more than half of M7 AML cases express CD42b. Most cases are positive for CD4 and CD33; CD13, CD34, CD36, CD45, and HLA-DR are infrequently detected.79 The term “M0” denotes minimally differentiated myeloid leukemia.92 In general, the expression of CD3, CD79α, or TCR proteins is strongly indicative of lymphoid lineage differentiation. In the absence of these lymphoid markers and of markers associated with the megakaryocytic lineage, the expression of CD13, CD15, CD33, CD65, or MPO is evidence of myeloid lineage commitment.79 Leukemias that are devoid of detectable MPO should be classified as M0 AML only in the absence of lineage-restricted lymphoid and megakaryocytic antigens.79 Although the cells of most M0 cases express CD13 or CD33, some MPO+ cases may lack these antigens.79 The expression of CD117 by leukemic cells is strongly suggestive of AML. Other non–lineage-restricted antigens found on M0 AML cells include CD2, CD4, CD7, CD9, CD10, CD11b, CD19, CD34, CD71, TdT, and HLA-DR.79
Childhood Leukemia
Introduction
Epidemiology
Congenital Disorder
Associated Leukemia(s)
Down syndrome
ALL, AML
Ataxia-telangiectasia
ALL
Wiskott-Aldrich syndrome
AML
Bloom syndrome
ALL, AML
Fanconi anemia
AML
Kostmann disease
AML
Neurofibromatosis
AML, JMML
Noonan syndrome
JMML
Etiology
Pathogenesis
General Clinical and Laboratory Features
Differential Diagnosis
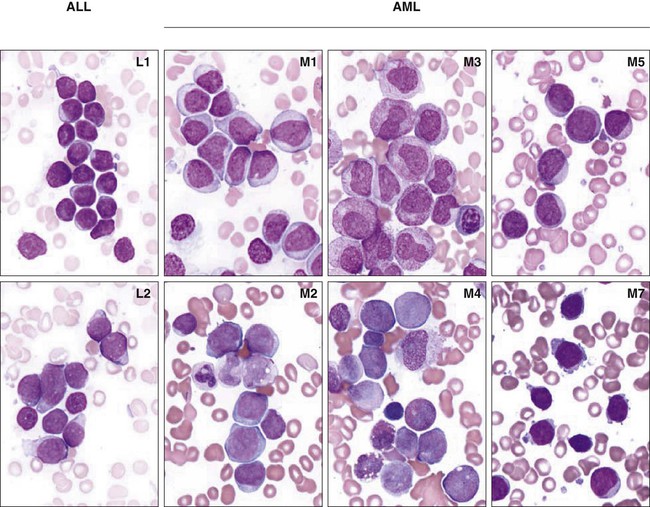
Morphologic and Cytochemical Analysis
Myelodysplastic/Myeloproliferative Disease
Down Syndrome Disease
MDS
JMML
Transient abnormal myelopoiesis
Refractory cytopenia (<2% blasts in blood and <5% blasts in marrow)
Chronic myelomonocytic leukemia (secondary to previous chemotherapy)
Myeloid leukemia of Down Syndrome
RAEB (2% to 19% blasts in blood or 5% to 19% blasts in marrow)
Ph-negative CML
RAEB-T (20% to 29% blasts in blood or marrow)
Immunologic Classification of Acute Leukemia
Acute Lymphoblastic Leukemia
Subtype
Antigen Expression (% of Cases Positive)
Frequency
CD19
cCD22
CD79α
CD10
CD7
CD5
cCD3
cIg µ
sIg µ
sIg κ or λ
Early pre-B
100
>95*
>95
95
5
0
0
0
0
0
60% to 65%
Pre-B
100
100*
100
>95
0
<2
0
100
0
0
20% to 25%
B
100
100*
100
50
0
0
0
>95
>95
>95
2% to 3%
T
<5
0
30
45
100
95
100a
0
0
0
15% to 18%
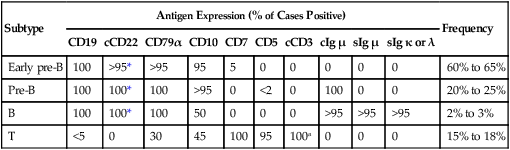
Early Pre-B Acute Lymphoblastic Leukemia
Pre-B Acute Lymphoblastic Leukemia
B-Cell Acute Lymphoblastic Leukemia
T-Lineage Acute Lymphoblastic Leukemia
Acute Myeloid Leukemia
FAB Subtype
Antigen Expression (Approximate % of Cases Positive for Marker)
CD34
CD117
HLA-DR
MPO
CD13
CD33
CD15
CD65
CD14
GPA
CD36
CD41a
M0
75
75
75
>80
75
75
30
30
0
0
0
0
M1
75
75
75
>80
75
75
75
75
0
0
0
0
M2
75
75
>80
>80
>80
>80
75
75
0
0
0
0
M3
<10
30
<10
>80
>80
>80
75
75
0
0
0
0
M4
75
75
>80
>80
75
>80
75
>80
75
0
30
0
M5
<10
30
>80
>80
75
>80
75
>80
75
0
75
0
M6
30
30
75
>80
75
75
30
75
0
>80
75
0
M7
30
30
30
0
30
75
30
<10
0
<10
75
>80
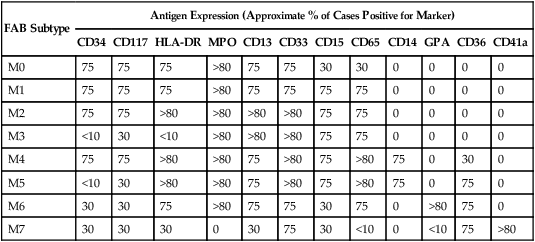
Acute Myelocytic Leukemia with Differentiation (M2 AML)
Acute Promyelocytic Leukemia (M3 AML)
Acute Myelomonocytic Leukemia (M4 AML)
Acute Monocytic Leukemia (M5 AML)
Acute Erythroleukemia (M6 AML) and Acute Erythroblastic Leukemia
Acute Megakaryoblastic Leukemia (M7 AML)
Acute Myeloid Leukemia Without Morphologic or Cytochemical Evidence of Differentiation (M0 AML)
Childhood Leukemia