Chest wall tumors are a rare collection of benign and malignant tumors involving the various layers of the thorax. Primary chest wall tumors constitute approximately 1% of all tumors. Of chest wall tumors, approximately half are benign, and half malignant. Management varies based on tumor type and risk of malignancy. Given the rare nature of chest wall tumors, formal guidelines for their management are lacking. However, review of past management practice as well as ongoing experience in chest wall tumor management has resulted in cumulative experience with which we approach chest wall tumors.
The incidence of different chest wall tumor types is variously reported in a limited number of case series. We have reported estimated incidence of more commonly occurring chest wall tumors based on the currently available series from the past three decades.1–5 Estimating incidence has been further complicated by reclassification of different tumors over the decades. As a result, the true incidence of chest wall tumors, as currently defined, is unclear.
Regarding the periodic and ongoing reclassification of chest wall tumor types, we have identified the following situations which the reader should be aware of. Fibrosarcoma was the most common soft tissue sarcoma (STS) reported in the first half of the 20th century.6 A review of fibrosarcomas by Stout in 1948 revealed that many tumors classified as such were actually rhabdomyosarcomas, liposarcomas, and synovial sarcomas.7 Malignant fibrous histiocytoma (MFH) was subsequently the most common soft tissue tumor type in adults starting in the 1960s. Modern pathologic review of MFH series reveal that many tumors previously identified as MFH have been reclassified as pleomorphic sarcomas originating from different tissue types.8 MFH is now renamed undifferentiated pleomorphic sarcoma and accounts for a much smaller percentage of STS than previously thought.9 Similarly, desmoid tumor was previously classified as low-grade fibrosarcoma but is now widely regarded as borderline malignant tumor with no metastatic potential.6 Reported series of primary chest wall tumors necessarily suffer from this same pathologic confusion as they have been collected over decades owing to the rarity of these tumors. In estimating the incidence of primary chest wall tumors we attempt to use modern pathologic designations whenever possible (Table 63-1).
Most Commonly Occurring Primary Chest Wall Tumors
| Benign |
| Fibrous Dysplasia |
| Chondroma |
| Osteochondroma |
| Aneurysmal bone cyst |
| Langerhans cell histiocytosis |
| Malignant |
| Soft tissue sarcoma |
| Rhabdomyosarcoma |
| Liposarcoma |
| Malignant peripheral nerve sheath tumor |
| Pleomorphic sarcoma |
| Fibrosarcoma |
| Synovial sarcoma |
| Solitary fibrous tumor |
| Alveolar soft part sarcoma |
| Dermatofibrosarcoma protuberans |
| Leiomyosarcoma |
| Desmoid |
| Chondrosarcoma |
| Ewing sarcoma |
| Osteosarcoma |
| Solitary plasmacytoma of bone |
Benign and malignant tumors each constitute approximately 50% of primary chest wall tumors.3,10 It is estimated that half of all malignant tumors of the chest wall represent secondary chest wall tumors (Table 63-2).11 These tumors are metastatic from other locations or, more commonly, arise by direct extension of an adjacent malignancy. Lung and breast cancer represent the most common malignancies directly invading the chest. About 5% of non-small cell lung cancers that are resected exhibit chest wall invasion. Resection of this group can provide a durable 5-year survival of 50%.12 Surgical treatment for locally recurrent breast cancer invading the chest wall should be evaluated individually as metastatic disease is likely present. Predictors of local recurrence are presence of nodal disease and tumor size greater than 4 cm. Predictors of poor long-term survival are presence of nodal disease and triple-negative phenotype (estrogen receptor (−), progesterone receptor (−), and Her2/Neu (−)). Five-year survival for this group is 18%.13,14 Renal cell carcinoma does not infrequently metastasize to the bony thorax and should be resected if all metastatic disease can be removed. Less common secondary chest wall tumors arise from metastatic colorectal, thyroid, prostate, or ovarian cancer.11,15
It is the aim of this review to provide the surgical oncologist with an overview of the most common chest wall tumors, and management guidelines. Given that rare chest wall tumors present with similar symptoms and findings to more common benign and malignant chest wall tumors, the authors recommend that a multidisciplinary review be performed of the less common tumors to ensure individual patient management is optimized.
Management of chest wall tumors is dictated by suspected or confirmed diagnosis. Given the rarity of most of these tumors, we advocate review of unusual cases by a multidisciplinary team of practitioners, each bringing their expertise and experience to bear. A multidisciplinary team should include thoracic surgeons, medical oncologists, radiation oncologists, radiologists, and pathologists, as well as social workers, nutritionists, and physical and occupational therapists. While surgical extirpation of tumors is a common approach, some tumor types benefit from chemotherapy and radiotherapy in the neoadjuvant or adjuvant setting. Post-treatment follow-up planning is also a critical component of chest wall tumor management.
The aim of surgical management of chest wall tumors is en bloc resection achieving an R0 resection. With modern surgical techniques very large, full thickness chest wall defects can be satisfactorily reconstructed in most cases. This often necessitates a multidisciplinary surgical approach involving cooperation between plastic and reconstructive surgeons, general surgeons, and in certain cases orthopedic and neurosurgeons. Reoperation for local recurrence can result in increased morbidity and increase the challenges of a subsequent chest wall reconstruction.
The desired chest wall reconstruction restores physiologic function by obviating paradoxical movement with respiration and providing stability for upper limb movement.16 In addition, exposure of vital organs and structures such as lung, heart, and major vessels necessitates coverage in order to provide sufficient protection. Finally, the cosmetic outcome must be considered when reconstructing resultant deformities.
In general, anterior and lateral chest wall defects less than 5 cm and superior posterior defects less than 10 cm occurring above the tip of the scapula do not need to be reconstructed and will not result in appreciable functional decline.17,18 Resection of up to two ribs, preserving overlying soft tissue, can often be reconstructed with local advancement of soft tissue to provide coverage. Larger anterior and lateral resections involving three ribs or more, full-thickness resections, posterior defects below the scapular tip, and complete sternectomy benefit from reconstruction.
Additional circumstances must be taken into consideration when performing chest wall resection and reconstruction. Sternal resection necessitates reconstruction to provide coverage for underlying structures and to restore chest wall stability in the setting of a complete sternectomy.19 If the chest wall ring remains intact, even at a single location, sternal reconstruction with a muscle flap is often satisfactory.16,19 When the chest wall defect is sufficiently large that a flail segment is likely to result, rigid or semirigid reconstruction should be performed. Posterior defects which underlie the area of excursion of the scapular tip should be reconstructed to prevent entrapment of it. The authors reconstruct anterior and lateral chest wall defects involving the lower ribcage with flexible material such as Gore-Tex (W. L. Gore & Associates, Inc., Newark, Delaware, USA), polytetrafluoroethylene (PTFE), or polypropylene mesh as this is better tolerated by the patient than rigid prostheses while bending over. When sewn with moderate tension into a chest wall defect, 2-mm Gore-Tex provides reasonable protection to underlying viscera, and provides a semirigid substrate which is comfortable and accommodates respiration and movement. Gore-Tex is smooth, air and liquid impermeable, and does not adhere to underlying viscera and therefore is unlikely to erode into it.
Rigid reconstruction is commonly accomplished with a mesh/resin sandwich17 or metal bars or plates used with or without mesh.19,20 The mesh/resin sandwich is constructed using methyl methacrylate sandwiched between two pieces of nonabsorbable mesh sized to the defect, and sewn in place with nonabsorbable suture. Metal reconstruction devices are titanium struts often custom sized to patients to span the resulting defect and affixed to remaining ribs or sternum (Strasbourg Thoracic Osteosyntheses System—STRATOS™; MedXpert GmbH, Heitersheim, Germany). Autologous rigid reconstruction can be accomplished by free rib transposition, using a rib remote from the surgical site.21 Semirigid moldable implants have been employed particularly for sternal reconstruction.22
When employing rigid material for chest wall reconstruction, care should be taken not to place this material in areas of a large degree of chest wall excursion. For example, mesh-methyl methacrylate sandwich may erode into the trachea when used for manubrial reconstruction. Similarly, use of mesh-methyl methacrylate sandwich for inferior, anterior chest wall defects can result in discomfort when patients bend forward. Therefore, consideration must be made when selecting material for chest wall reconstruction.
Partial thickness soft tissue defects of the chest wall can usually be covered with a local tissue advancement flap or a skin graft. Options for full thickness chest wall reconstruction include use of nonrigid or rigid materials followed by soft tissue coverage with a local flap or free flap. In situations where placement of foreign material is contraindicated, advanced autologous tissue transfer techniques are useful. These situations include infection and gross contamination from enteral visceral. Free tissue flaps can be used in most locations but are generally reserved for situations where regional flaps are unavailable, and include bone, myocutaneous, and cutaneous tissue transfer.23 Transposition of previously radiated muscles greatly increases the risk of total muscle necrosis and should be avoided if possible.24
Autogenous tissue transfer options include local tissue advancement flaps, rotational flaps, and free tissue flaps. Small defects can typically be covered using advancement of local tissues.24,25 The latissimus dorsi muscle superiorly pedicled off of the thoracodorsal branch of the subscapular artery can be used for coverage of lateral and anterior defects.16,24,26 The pectoralis major muscle pedicled laterally off of the pectoral branch of the thoracoacromial artery can be used for coverage of anterior and lateral defects, including those resulting from manubrial and sternal resection. The pectoralis major muscle pedicled medially off of perforating arteries from the internal mammary artery can be used for coverage of contralateral anterior defects, including those resulting from manubrial and sternal resection. The serratus anterior muscle pedicled superiorly off of the lateral thoracic artery can be used for coverage of posterior defects. The trapezius muscle can cover medial defects of the upper back. In addition, omental or rectus abdominis flaps can also be used to fill chest wall defects.16,24 Omental flaps are particularly useful for filling contaminated wounds owing to its extensive blood supply and network of lymphatics. It is useful for providing a matrix between prosthetic reconstruction and an overlying skin graft.16
Radiotherapy to the chest wall is commonly done for breast cancer and is also used in treating primary chest wall tumors and lung cancer. Thoracic radionecrosis can require debridement including full thickness chest wall resection. These necrotic lesions are frequently infected and should be reconstructed with well-vascularized autologous tissue.
Complications from chest wall resections and reconstructions are not uncommon. A 46% to 69% overall complication rate and a 24% to 27% respiratory complication rate has been reported following large defect reconstruction of more than two ribs or the sternum.17,18,20 Other common complications following these operations include infection, hematoma, seroma formation, and pleuroparenchymal fistulae.
Biologic mesh is useful in the setting of wound infection or contamination.27 Wound infection complicates approximately 5% of chest wall reconstructions.18 Within a few weeks of prosthetic chest wall reconstruction, a fibrous capsule envelops the prosthesis. In the setting of wound infection, nonabsorbable prosthetic material should be removed. Should this occur prior to stabilization of the chest wall defect with fibrous capsule development, a combination of absorbable mesh and autogenous tissue transfer may be used to fill the defect. If the chest wall has already stabilized, removal of the infected prosthesis and healing by secondary intent may suffice.
Soft tissue sarcomas are a rare and diverse group of neoplasms of mesenchymal origin. As such they arise in muscle, fat, blood vessels, and other fibrous tissue and can be present anywhere in the body. STS represent less than 1% of all solid tumors with 5% to 15% of these occurring in the chest wall.9,28,29
The cause of STS is unknown with most of these tumors arising de novo. Some congenital syndromes have been associated with STS including Li–Fraumeni (rhabdomyosarcoma), neurofibromatosis (malignant peripheral nerve sheath tumors), and hereditary retinoblastoma (STS). Environmental exposures including phenoxyacetic acids and chlorophenols30 have been shown to increase the risk of STS. Postirradiation sarcomas have been observed particularly in breast cancer patients treated with adjuvant radiotherapy where the risk of developing STS rises with increasing energy dose.31 Finally, human herpes virus 8 and Epstein–Barr virus have been associated with Kaposi sarcoma and leiomyosarcoma respectively in the immunosuppressed patient.32
These tumors usually present as a slow-growing painless mass (Fig. 63-1). As they increase in size, however, they may exert a local compressive effect causing pain, paresthesias, and swelling. This can be particularly true for tumors involving the shoulder girdle where large tumors can compress major nerves and vessels. Growth rates are variable with a higher rate of growth associated with a higher tumor grade and increased mortality.33
Magnetic resonance imaging (MRI) is our modality of choice for determining tumor size and characterizing the relationship of the tumor to bone and nearby neurovascular structures. Computed tomography (CT) of the chest is used for evaluating the chest for metastatic disease. Enlarged lymph nodes can be evaluated by biopsy (percutaneous, endobronchially, or via mediastinoscopy) or positron emission tomography/CT (PET/CT).
Tissue diagnosis is essential to establish a diagnosis, and grade the tumor. Enough tissue must be obtained to adequately assess the mass. Core needle biopsy is now frequently adequate though the historic standard of incisional or excisional biopsy is occasionally necessary to obtain sufficient tissue to establish tumor histology and grade.34 The biopsy site should be oriented with future surgery in mind so that any needle tract or biopsy site is excised with the surgical specimen.
Chest wall STS behaves clinically most like extremity sarcomas where the risk of local recurrence is most strongly predicted by inadequate resection. The risk of metastasis is associated with tumor grade.35,36 The primary therapy for chest wall STS is wide local resection with margins greater than 1 cm. For high-grade tumors treatment should be guided by size and ability to achieve an adequate resection. In the case of larger, potentially resectable tumors neoadjuvant radiotherapy, chemotherapy, or chemoradiotherapy may be offered. Adjuvant radiotherapy should be considered for positive or margins less than 1 cm.
The optimal chemotherapy regimen for STS has not been defined, however multiple agents have shown some benefit in combination with radiotherapy including doxorubicin, ifosfamide, gemcitabine, and temozolomide. Though a survival benefit has yet to be shown, some authors advocate multiagent chemotherapy over single agent for large STS and have demonstrated improved disease-specific survival.37
Data on recurrence rates of chest wall STS are limited; however several series suggest a recurrence rate of 20% to 25%. Local recurrences occur between 9% and 11%. Undifferentiated pleomorphic sarcoma represents the most common histologic subtype associated with local recurrence. The most common site of distant recurrence is the lungs. The majority of recurrences occur within the first 3 years after initial treatment and are optimally treated with re-excision. Local recurrences are frequently symptomatic and evident on physical exam in contrast to distant recurrences which are usually found on follow-up screening. Therefore, regular screening with chest CT and frequent physical exam after resection of a chest wall STS is prudent.35 We obtain a postoperative chest CT or MRI (depending on the multidisciplinary team thoracic radiologist’s preference) 3 months after resection, then yearly thereafter.
Liposarcoma is an adipocyte tumor encompassing multiple variants. Well-differentiated liposarcoma, also known as atypical lipomatous tumor, represents 40% to 45% of this category and is characterized by its inability to metastasize. Microscopically, these tumors reveal mature adipocytes with atypical nuclei with scattered fibrous tissue and few mitotic figures. They commonly reveal amplification of the MDM2 gene found on chromosome 12.9 MRI reveals signal intensity similar to fat with low signal intensity on T1-weighted images and high intensity on T2-weighted images.38 They occur most commonly in the extremities and retroperitoneum and only rarely in the chest wall.39 When found in the chest wall, they can be subcutaneous or in the intermuscular fascia.40 The site of occurrence is a primary predictor of survival with central body sites having a worse prognosis due to the difficulty in achieving adequate wide resection. Thus, the rate of local recurrence is much higher in these cases. When local recurrence is observed, a proportion of well-differentiated liposarcomas recur as dedifferentiated liposarcoma in which case metastases can occur and prognosis is further worsened.9,39
Dedifferentiated liposarcoma, though occurring in 10% of well-differentiated liposarcomas, most often arises de novo.9 Nearly 70% of these tumors arise in the retroperitoneum with less than 10% arising in the trunk region.41 A rapid increase in the rate of growth of a previously present mass can signal the regression of well-differentiated liposarcoma to dedifferentiated liposarcoma. Distant metastases occur in 17% of cases and the 5-year survival is 72%.41
Myxoid liposarcoma is the second most common subtype of liposarcoma composing 30% of cases.9 It frequently occurs in younger patients and arises in lower extremities. It metastasizes more frequently to bone than the lungs. Ninety-five percent of cases of myxoid liposarcoma are marked by the balanced translocation t(12;16)(q13;p11) and fusion of the CHOP/DDIT3 gene with the FUS gene.42
Pleomorphic liposarcoma is the least common of liposarcoma subtypes accounting for 5% of cases. It is characterized by pleomorphic lipoblasts amidst high-grade, pleomorphic sarcoma. No reliable genetic marker has been identified for this tumor subtype.9,42 About half of cases occur in lower extremities followed in frequency by the upper extremity and trunk. Local recurrence and metastases are each seen in about 30% of patients.43
Treatment of liposarcoma, as per that of most STS, is margin-negative resection. Radiotherapy may be utilized for positive margins and recurrence when surgery is not feasible.
Rhabdomyosarcoma (RMS) is the most common pediatric STS accounting for almost 50% in children age 0 to 14 years. Overall, however, RMS is a rare tumor constituting only 3.1% of STS.44 Case series suggest RMS accounts for 21% of STS of the chest wall.2
The diagnosis of RMS is achieved by identifying skeletal muscle lineage in specimens. This requires examination by light microscopy and immunohistochemical evaluation for desmin, actin, myoglobin, and myogenin. The most recent World Health Organization (WHO) classification divides RMS into four subtypes: embryonal (the most common subtype in children and adolescents); alveolar (48% of chest wall RMS); pleomorphic (the most common subtype in the elderly); and spindle cell.9,45 Genetic analysis reveals loss of heterozygosity for chromosome 11 in most cases of embryonal RMS causing tumor suppressor gene inactivation. Alveolar RMS is characterized by consistent chromosomal translocations resulting in transcription factor activation.9
Rhabdomyosarcoma of the chest wall is typically a fast growing tumor that presents as a mass, either asymptomatic or painful. One case series found the median tumor size to be 7 cm on presentation with 20% having stage IV disease at diagnosis.46 This highlights the aggressive nature of this tumor. Staging workup involves MRI to evaluate the extent of the primary tumor and PET/CT to evaluate for metastatic disease.
Surgical excision remains the mainstay of treatment for localized chest wall RMS. It is important to evaluate resectability prior to attempting resection. If positive margins are likely then neoadjuvant chemotherapy should be considered with the aim of decreasing the tumor burden and facilitating achievement of an R0 resection. Microscopically positive margins are treated with radiotherapy and do not result in decreased survival compared with negative margins.45
Malignant peripheral nerve sheath tumors (MPNST) account for 5% to 10% of all STS and compose 9% of all chest wall STS.2,9,47 Twenty percent to 50% of all MPNST arise in the setting of the hereditary syndrome neurofibromatosis 1 (NF1) and carry a worse prognosis than sporadic MPNST.9,39 Ten percent are associated with previous chest wall radiotherapy.9 This tumor most commonly occurs in the extremities and involves the thorax in 15% of cases.39
Typical MPNST shows fascicles of spindle cells with hyperchromatic nuclei which are mitotically active.9,47 Many MPNST tumors exhibit mutations of the NF1 gene encoding the tumor suppressor neurofibromin. Other complex changes in karyotype can be seen in MPNST with no clear difference between NF1-associated and sporadic MPNST tumors.9
Malignant peripheral nerve sheath tumor is an aggressive tumor with poor survival with prognosis linked to location on the trunk, increased size, association with NF1, and increased grade of tumor. Five-year survival for NF1-associated MPNST is 16% to 38% and for sporadic MPNST is 42% to 57%.39,47
Chest wall MPNST present as a rapidly enlarging asymptomatic or painful chest wall mass. Neurologic symptoms, pleuritic pain, and dyspnea may occur as it enlarges.48 CT imaging often reveals bone destruction in the setting of a heterogenous mass. On MRI, the tumor exists along the course of a peripheral nerve and is heterogenous and irregular with increased signal intensity on T2-weighted imaging.38 Invasion of fat planes and surrounding edema is also often present.49 When MPNST is suspected open biopsy should be used to confirm the diagnosis.50,51
The treatment of MPNST requires a multimodal approach with a focus on a microscopically margin-negative surgical resection. Adjuvant radiotherapy and chemotherapy are administered for large tumors greater than 5 cm, high-grade tumors, and positive margins.47
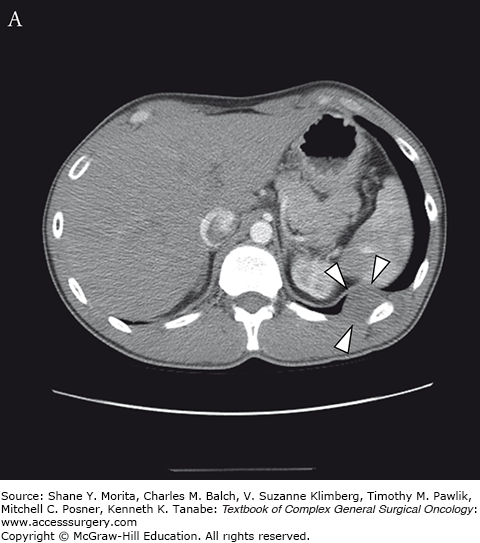
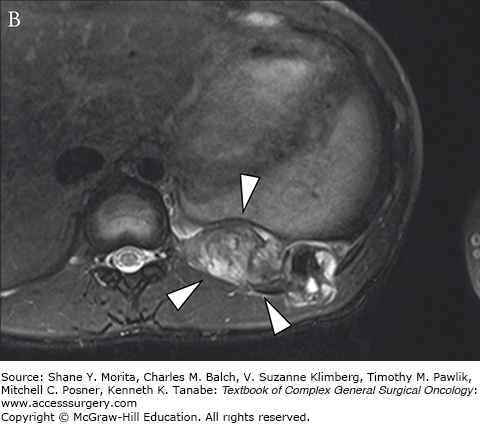
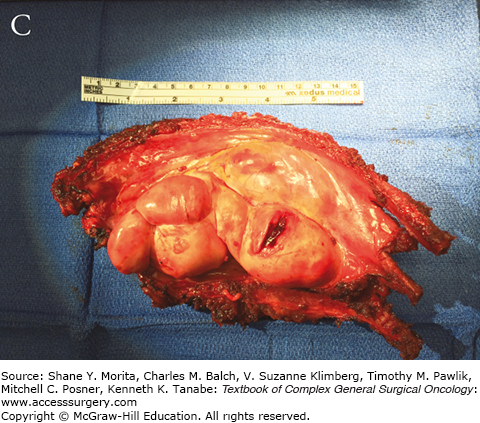
The group of soft tissue tumors termed pleomorphic sarcomas, and formerly known as malignant fibrous histiocytoma, is the most common STS in adults. Occurring most frequently in the extremities and retroperitoneum, it is a rare primary chest wall tumor with an overall incidence of 1 to 2 per 100,000.9 This tumor presents as a rapidly growing mass which may cause pain as it enlarges. It most often involves deep fascia or skeletal muscle and has been observed to cause bony erosion or invasion of ribs (Fig. 63-2).52,53 Metastases are present at the time of diagnosis in 5% of cases, and most frequently involve the lungs. The genetic basis for this tumor has not yet been characterized.
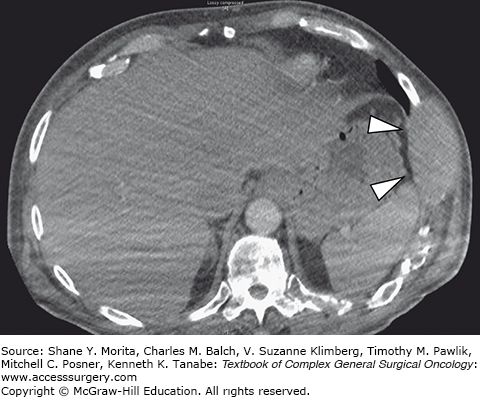
Grossly these tumors are heterogeneous with a variable cut surface. Fleshy, cystic, and necrotic areas can all be present in the same tumor. These findings are reflected on imaging where MRI reveals a homogeneous or heterogenous mass with a T1 signal intensity equal to muscle. T2-weighted images show an intensity equal to or greater than fat.38
Pleomorphic sarcoma is an aggressive tumor with an overall 5-year survival of 60% to 70%.54 There is variation in prognosis among the subgroups of this tumor classification.
Fibrosarcoma (FS) has been reported to account for 11% to 18% of STS of the chest wall.2,4 Though previously considered the most common adult sarcoma, the rates of FS have declined dramatically in recent years under the modern classification system for STS.6 Therefore, the historically reported rate of FS of the chest wall is likely an overestimate.
Fibrosarcoma is characterized by fascicles existing in a herringbone pattern with scant cytoplasm and variable mitotic activity.6 Various chromosomal abnormalities have been identified in FS without a single common genetic mutation.9 A recently reexamined series of FS patients using the current classification system reveals the aggressive nature of this tumor. High-grade tumors composed 85% of the cohort. A high rate of local recurrence and metastasis were observed and overall 5-year survival was less than 55%. This tumor occurs most commonly in an older population (median age 55 years) and in the lower extremities 46% of the time. Only 7% of patients in this cohort had a chest wall fibrosarcoma.55
Like other STS a microscopically margin-negative surgical resection should be achieved to reduce the high rates of local recurrence commonly seen with this tumor. Prognostic variables for FS from previous data are histologic grade, size of primary tumor, and depth of invasion.9
Synovial sarcoma accounts for 5% of STS, most of which occur before the age of 50 and more frequently in males.56 These tumors can occur anywhere but most commonly arise in soft tissue near joints or tendon sheaths in the extremities.9 Multiple case reports of chest wall synovial sarcomas reveal their ability to invade virtually any surrounding structure including vessels, nerves, ribs, pericardium, and diaphragm.57–59 They tend to grow slowly and create symptoms based on the structures they compress and invade.
Computed tomography imaging may reveal calcifications and evidence of bone destruction. On MRI, the tumor is well defined with multiple loculations. Internal hemorrhage is a common characteristic of synovial sarcomas and is represented by high signal intensity on both T1- and T2-weighted images. T2-weighted images show marked heterogeneity owing to the cystic and solid elements of this tumor.60
Greater than 90% of these tumors show translocation (X;18)(p11;q11) on genetic analysis, and is a key to diagnosis of synovial sarcoma.9 This high-grade tumor conveys a poor prognosis with historical series suggesting a local recurrence rate of 50%. Treatment is microscopically margin-negative surgical resection. Adjuvant radiotherapy has shown improvement in local recurrence rates. However distant metastases, mostly to the lungs and bone, are still seen in 40% of cases.61
Solitary fibrous tumor (SFT), previously termed hemangiopericytoma or pleural fibroma, is a rare mesenchymal tumor most commonly occurring in middle-aged adults and seldom encountered in children and adolescents.9 SFT has classically been described as a well-circumscribed, pleural-based tumor. However, it is now evident that in 30% to 40% of cases, SFT occurs in extrapleural locations.62, 63 In case series, SFT account for less than 1% to 3% of primary chest wall tumors, with only about 800 cases reported to date.2–4,64
Solitary fibrous tumor originate in the submesothelial mesenchymal layer of the pleura. Malignant extrapleural SFT appears as well circumscribed and multilobulated with infiltrative borders, necrosis, and increased mitotic activity (greater than 4 per 10 high power fields (HPF)). The majority of SFTs express CD34 and contain the gene fusion NAB2-STAT6.64,65 Overexpression of P53 has been observed in the most high-grade portions of this tumor.9
Solitary fibrous tumor commonly exhibits foci of low signal intensity on both T1- and T2-weighted MRI. It enhances on both CT and MRI. This produces a “chocolate chip cookie” appearance which may aid in diagnosis. FDG avidity on PET is seen with malignant SFT.66
Patients are most commonly asymptomatic, but cough, chest pain, and dyspnea are present in 8% to 33% of cases. SFT is associated with paraneoplastic syndromes, including hypertrophic pulmonary osteoarthropathy (22%) and symptomatic hypoglycemia (4%). Patients with hypertrophic pulmonary osteoarthropathy present with bilateral arthritic-like symptoms of the lower extremity long bones.64
Margin-negative surgical resection is the cornerstone of curative treatment—this tumor responds poorly to chemotherapy.9,63 Recurrence is increased when malignant histology, benign histology with atypia, and extrathoracic tumor location are present. Tumor size greater than 10 cm and high mitotic activity have also been associated with an increased rate of metastases.9,67 Some benign SFT recur up to 17 years later with the recurrence containing more malignant histology. With resection of malignant SFT, 50% of patients will be cured. However, 25% of patients will develop widespread disease. The local recurrence rate is 8%and most patients with recurrence will die within 2 years.63,64 Given the risk of recurrence and metastasis of “benign” SFT, the authors raises the question of whether these tumors can be considered benign versus malignant. Instead, we treat these tumors clinically as low- versus high-grade malignancy, and approach all with the aim of surgical cure with wide negative margins.
Alveolar soft part sarcoma (ASPS) is a rare tumor with a frequency up to 0.9% of all STS.9 In case series of primary malignant chest wall tumors, ASPS is similarly rare with a reported frequency of 0.8%.2 This tumor occurs two times more frequently in women and most frequently in patients aged 15 to 35 years.9 In adults, ASPS occurs most frequently in the deep tissues of the lower extremities, particularly the thigh, and in children occurs most commonly in the head and neck region.68
This tumor is typically poorly circumscribed with areas of necrosis and hemorrhage. The origin of this tumor is unknown though some evidence suggests skeletal muscle differentiation.69 On gross appearance the tumor is poorly circumscribed with areas of hemorrhage and necrosis.9 Histology reveals an organoid or alveolar pattern separated by vascular channels. The abundant cytoplasm is granular and eosinophilic.9,69 Strong nuclear staining for TFE3 is consistently found in most ASPS tumors. Identification of a chromosomal translocation resulting in the ASPSCR1-TFE3 fusion is highly sensitive and specific for ASPS.9
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


