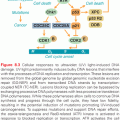
Genotoxic carcinogens may transfer simple alkyl or complexed (aryl) alkyl groups to specific sites on DNA bases.18,21 These alkylating and aryl-alkylating agents include, but are not limited to, N-nitroso compounds, aliphatic epoxides, aflatoxins, mustards, polycyclic aromatic hydrocarbons, and other combustion products of fossil fuels and vegetable matter. Others transfer arylamine residues to DNA, as exemplified by aryl aromatic amines, aminoazo dyes, and heterocyclic aromatic amines. For genotoxic carcinogens, the interaction with DNA is not random, and each class of agents reacts selectively with purine and pyrimidine targets.7,18,21 Furthermore, targeting carcinogens to particular sites in DNA is determined by nucleotide sequence, by host cell, and by selective DNA repair processes (see later discussion), making some genetic material at risk over others. As expected from this chemistry, genotoxic carcinogens can be potent mutagens and particularly adept at causing nucleotide base mispairing or small deletions, leading to missense or nonsense mutations. Others may cause macrogenetic damage, such as chromosome breaks and large deletions. In some cases, such genotoxic damage may result in changes in transcription and translation that affect protein levels or function, which in turn alter the behavior of the specific host cell type. For example, there may be effects on cell proliferation, programmed cell death, or DNA repair. This is best typified by the signature mutations detected in the p53 gene caused by ingested aflatoxin in human liver cancer22 and by polycyclic aromatic hydrocarbons human lung cancer caused by the inhalation of cigarette smoke.15,23,24 Similarly, a distinct pattern of mutations is detected in pancreatic cancers from smokers when compared with pancreatic cancers from nonsmokers.25
Some chemicals that cause cancers in laboratory rodents are not demonstrably genotoxic. In general, these agents are carcinogenic in laboratory animals at high doses and require prolonged exposure. Synthetic pesticides and herbicides fall within this group, as do a number of natural products that are ingested. The mechanism of action by nongenotoxic carcinogens is not well understood, and may be related in some cases to toxic cell death and regenerative hyperplasia. They may also induce endogenous mutagenic mechanisms through the production of free radicals, increasing rates of depurination, and the deamination of 5-methylcytosine. In other cases, nongenotoxic carcinogens may have hormonal effects on hormone-dependent tissues. For example, some pesticides, herbicides, and fungicides have endocrine-disrupting properties in experimental models, although the relation to human cancer risk is unknown.
ANIMAL MODEL SYSTEMS AND CHEMICAL CARCINOGENESIS
Most human chemical carcinogens can induce tumors in experimental animals; however, the tumors may not be in the same organ, the exposure pathways may differ from human exposure, and the causative mechanisms may not exist in humans. In many cases, however, the cell of origin, morphogenesis, phenotypic markers, and genetic alterations are qualitatively identical to corresponding human cancers. Furthermore, animal models have revealed the constancy of carcinogen–host interaction among mammalian species by reproducing organ-specific cancers in animals with chemicals identified as human carcinogens, such as coal tar and squamous cell carcinomas, vinyl chloride and hepatic angiosarcomas, aflatoxin and hepatocellular carcinoma, and aromatic amines and bladder cancer. The introduction of genetically modified mice designed to reproduce specific human cancer syndromes and precancer models has accelerated both the understanding of the contributions of chemicals to cancer causation and the identification of potential exogenous carcinogens.26,27 Furthermore, construction of mouse strains genetically altered to express human drug–metabolizing enzymes has added both to the relevance of mouse studies for understanding human carcinogen metabolism and the prediction of genotoxicity from suspected human carcinogens and other chemical exposures.28 Together, these studies have indicated that carcinogenic agents can directly activate oncogenes, inactivate tumor suppressor genes, and cause the genomic changes that are associated with autonomous growth, enhanced survival, and modified gene expression profiles that are required for the malignant phenotype.29
Genetic Susceptibility to Chemical Carcinogenesis in Experimental Animal Models
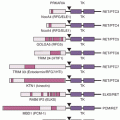
The use of inbred strains of rodents and spontaneous or genetically modified mutant strains have led to the identification and characterization of genes that modify risks for cancer development.30–32 For a variety of tissue sites, including the lungs, the liver, the breast, and the skin, pairs of inbred mice can differ by 100-fold in the risk for tumor development after carcinogen exposure. Genetically determined differences in the affinity for the aryl hydrocarbon hydroxylase (Ah) receptor or other differences in metabolic processing of carcinogens is one modifier that has a major impact on experimental and presumed human cancer risk.33–35 The development of mice reconstituted with components of the human carcinogen–metabolizing genome should facilitate the extrapolation of metabolic activity by human enzymes and cancer risk.27,28,36 Such mice also show that other loci regulate the growth of premalignant foci, the response to tumor promoters, the immune response to metastatic cells, and the basal proliferation rate of target cells.30 In mice susceptible to colon cancer due to a carcinogen-induced constitutive mutation in the APC gene, a locus on mouse chromosome 4 confers resistance to colon cancer.31 The identification of the phospholipase A2 gene at this locus and subsequent functional testing in transgenic mice revealed an interesting paracrine protective influence on tumor development.31 This gene, and several other genes mapped for susceptibility to chemically induced mouse tumors (PTPRJ, a receptor type tyrosine phosphatase, and STK6/STK15, an aurora kinase), have now been shown to influence susceptibility to organ-specific cancer induction in humans.30,31
MOLECULAR EPIDEMIOLOGY, CHEMICAL CARCINOGENESIS, AND CANCER RISK IN HUMAN POPULATIONS
Molecular epidemiology is the application of biologically based hypotheses using molecular and epidemiologic methods and measures. New technologies continue to allow epidemiologic studies to improve the testing of biologically based hypotheses and to develop large datasets for hypothesis generation, most notably the application of various –omics technologies via next-generation sequencing (e.g., genomics, epigenomics, transcriptomics), proteomics, and metabolomics. The greatest challenge now is to develop methods that allow for analysis cutting across various technologies.37–43 Recent advances now include the role of microRNA and long noncoding RNAs in tumor development and progression because of their impact on the regulation of gene expression.44,45 Chemical effects on microRNAs and the resultant gene expression is currently being identified.46 Using such technologies, emerging evidence is noting the importance of the microbiome and associated infections as a risk of human cancer.47–50 The complexity of environmental exposure and how it interacts with humans to affect numerous biologic pathways has been characterized as the exposome, also expressed as a multidimensional complex dataset.51 Therefore, the important goal remains: to characterize cancer risk based on gene–environment interactions. However, we remain challenged because cancer is a complex disease of diverse etiologies by multiple exposures causing damage in different genes; for example, genen–environmentn interactions, for which the variable n is not known.
Two fundamental principles underlie current studies of molecular epidemiology. First, carcinogenesis is a multistage process, and behind each stage are numerous genetic events that occur either due to an exogenous insult such as a chemical exposure or an endogenous insult, such as from free radicals generated via cellular processes or errors in DNA replication. Therefore, identifying a cancer risk factor can be challenging because of the multifactorial nature of carcinogenesis, given that any one risk factor occurs within a background of many risk factors. Second, wide interindividual variation in response to carcinogen exposure and other carcinogenic processes indicate that the human response is not homogeneous, so that experimental models and epidemiology (e.g., the use of a single cell clone to study a gene’s effect experimentally or the assumption that the population responds similarly to the mean in epidemiology studies), might not be representative of susceptible and resistant groups within a population.
Genetic Susceptibility
In humans, the determination of genetic susceptibility can be assessed by phenotyping or genotyping methods. Phenotypes generally represent complex genotypes. Examples of phenotypes include the assessment of DNA repair capacity in cultured blood cells, mammographic breast density, or the quantitation of carcinogen-DNA adducts in a target organ. Phenotypes now also include profiles of methylation that affect gene expression, a so-called epigenetic effect, for example, identified though next-generation sequencing or other methods.52 The contribution of genetics to cancer risk from chemical carcinogens can range from small to large, depending on its penetrance.4 Highly penetrant cancer-susceptibility genes cause familial cancers, but account for less than 5% of all cancers. Low-penetrant genes cause common sporadic cancers, which have large public health consequences.
A genetic polymorphism (e.g., single nucleotide polymorphisms) is defined as a genetic variant present in at least 1% of the population. Because of the advent of improved genotyping methods that have reduced cost and increased high throughput, haplotyping and whole genomewide association studies are ongoing. Although haplotyping studies, facilitated through the International HapMap Project (www.hapmap.org), have not proven useful for predicting human cancers; high-density, whole genomewide, single nucleotide polymorphism association studies have shown remarkable consistency for many gene loci, although the risk estimates are only 1.0 to 1.4, which are not useful in the clinic for individual risk assessment.6 For example, the contribution of genetic polymorphisms to cancer risk, at least for breast cancer, appears to improve risk modeling by only a few percent; known breast cancer risk factors account for about 58% of risk, and adding 10 genetic variants increases the risk prediction only to 62%.53 Genes under study are from pathways that affect behavior, activate and detoxify carcinogens, affect DNA repair, govern cell-cycle control, trigger apoptosis, effect cell signaling, and so forth.
Biomarkers of Cancer Risk
The evaluation of dose and risk estimates in epidemiologic studies can include four components: namely, external exposure measurements, internal exposure measurements, biomarkers estimating the biologically effective dose, and biomarkers of effect or harm. The latter three measurements are biomarkers that improve on the first by quantifying exposure inside the individual and at the cellular level to characterize low-dose exposures in low-risk populations, providing a relative contribution of individual chemical carcinogens from complex mixtures, and/or estimating total burden of a particular exposure where there are many sources.54
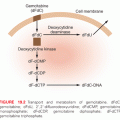
Chemicals cause genetic damage in different ways, namely in the formation of carcinogen-DNA adducts leading to base mutations or gross chromosomal changes. Adducts are formed when a mutagen, or part of it, irreversibly binds to DNA so that it can cause a base substitution, insertion, or deletion during DNA replication. Gross chromosomal mutations are chromosome breaks, gaps, or translocations. The level of DNA damage is the biologically effective dose in a target organ, and reflects the net result of carcinogen exposure, activation, lack of detoxification, lack of effective DNA repair, and lack of programmed cell death. A variety of assays have been used for determining carcinogen-macromolecular adducts in human tissues; for example, for assessing risk from tobacco smoking for lung cancer and aflatoxin and liver cancer.55,56 Important considerations for the assessment of biomarkers include sensitivity, specificity, reproducibility, accessibility for human use, and whether it represents a risk measured in a target organ or surrogate tissue. No single biomarker has been considered to be sufficiently validated for use as a cancer risk marker in an individual as it relates to chemical carcinogenesis.57 However, there is some evidence that DNA adducts are cancer risk factors in both cohort and case-control studies.58
People are commonly exposed to N-nitrosamine and other N-nitroso compounds from dietary and tobacco exposures, which are associated with DNA adduct formation and cancer. Exposure can occur through endogenous formation of N-nitrosamines from nitrates in food or directly from dietary sources, cosmetics, drugs, household commodities, and tobacco smoke. Endogenous formation occurs in the stomach from the reaction of nitrosatable amines and nitrate (used as a preservative), which is converted to nitrites by bacteria. The N
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


