Fig. 9.1
Main challenges associated with precision medicine trials
The main objective of PM designs is to identify molecular biomarkers that predict a clinical response (clinical validity) for MTA and to ultimately improve outcomes (clinical utility). PM trials might involve several tumor types, several molecular alterations, and MTAs, which represent a new challenge for biostatisticians. Several designs are today used for this purpose as detailed in chapter 8. Randomized prospective trials are important for patients’ follow-up, and data collection, otherwise PFS, is very difficult to interpret with respect to evaluating the effect of a treatment because the rate of PFS of different types of cancer varies substantially (Le Tourneau et al. 2014). An ideal design would determine a sample size in order to evaluate treatment effects with enough statistical power in any subgroup of patients with a specific tumor type harboring a specific molecular alteration and treated with a specific treatment. This setup would require thousands of patients and would therefore not be feasible in practice. New trial designs enriched for selected patients to receive a specific treatment, such as randomized discontinuation trials or adaptive randomization, are used today and allow controlling heterogeneity (refer to Chap. 8 for PM trial designs).
PM trials implicate numerous imperative stakeholders, including physicians, radiologists, pathologists, biostatisticians, high-throughput screening platforms managers, bioinformaticians, and biologists. The interactions of these different stakeholders need to be synchronized in order to be able to deliver the appropriate molecular information for patients’ treatment.
9.2 Setup of a Precision Medicine Trial: SHIVA Trial Implementation at Institut Curie
The setup and implementation of the SHIVA trial at Institut Curie end of 2012 may provide a realistic example of the challenges behind the setup of this new generation of clinical trials.
The SHIVA trial is the first randomized PM trial worldwide. SHIVA is a multicenter open-label randomized phase II trial involving patients with refractory cancer. In order to randomize and to treat 200 patients, up to 1000 patients were planned to be screened as explained below (Le Tourneau et al. 2014).
Patients meeting selection criteria sign first a consent form in order to perform a biopsy/resection of one metastasis and to establish a molecular profile of their tumor. If no molecular alteration for which an approved matched MTA exists according to a pre-specified treatment algorithm in the frame of the SHIVA trial is identified, patients are not eligible for the randomization and enter into a prospective observational cohort. If one or several molecular alterations are identified, the patient is given a second consent form to sign and has to meet the randomization criteria for randomization.
The setup of SHIVA trial at the French national level required a continuous contact with the clinical research operations, molecular and bioinformatics platforms, biostatisticians and researchers, as well as certified biologists. Several steps following patients’ consent necessitated the standardization of the different procedures:
9.2.1 Sample Collection
Standard operating procedures for tissue sampling and biobanking are mandatory for PM trials. In the frame of the SHIVA trial, three tumor samples were required for each patient. One biopsy was fixed and paraffin embedded for diagnostic confirmation as well as estrogen (ER), progesterone (PR), and androgen (AR) receptor expression analyses by IHC. The other biopsies were fresh frozen. One of them was used for DNA extraction using the kit Qiagen® after the evaluation of tumor cell content on a frozen section performed before extraction to allow microdissection of the sample to increase tumor cellularity (refer to Chap. 2 for pathology). Samples containing >30 % of tumor cells were considered suitable for DNA extractions and genomic analyses. This step was performed in all participating centers using a unique protocol. DNA quantity and quality were also assessed before transfer to the molecular platforms. The remaining frozen biopsies were used in case of insufficient DNA amount/quality for molecular analyses or otherwise stored for further studies.
9.2.2 Molecular Analyses
The use of high-throughput screening techniques in PM trial is not a trivial question since it includes a constant and complex interaction between different stakeholders and implicates a synchronized effort from tumor biopsy to result delivery. Several steps need to be standardized to ensure homogeneous procedures for all the patients within the trial and consequently guarantee a potential reproducibility of the results. This quite logical strategy is difficult to actually implement in the absence of clear guidelines for molecular analysis. The molecular profile of each patient enrolled in the SHIVA trial was performed using Ion Torrent/PGM (Life Technologies®) for mutation detection, CytoScan HD (Affymetrix®) for gene copy number alterations, and immunohistochemistry (IHC) for protein expression assessments and amplifications/deletion validation.
Molecular analyses were performed on three different platforms throughout France: Institut Curie, Paris, Centre Léon-Bérard, and Centre René Gauducheau, Nantes.
The majority of the molecular analyses were centralized at Institut Curie for the other recruiting centers. In order to ensure that all analyses were standardized, five DNA samples were processed at the same time on the different platforms for both mutation and gene copy number alteration analyses. The data were then collected and analyzed, and the reproducibility of the results was mandatory for the prospective analyses. In addition, two versions of the AmpliSeq cancer panel were used in the frame of the trial, and the transition to the second version also required a standardization procedure.
Concerning the hormone receptor expression levels, the technique is a routine procedure and consequently was performed by the different centers. For the validation of amplifications and deletions, the IHC was centralized at Institut Curie to ensure homogeneous results. For example, the genomic loss or a deletion of PTEN has been performed using a standard procedure for the SHIVA trial (refer to Chap. 2).
9.2.3 Bioinfomatics and Molecular Report
All NGS and CytoScan HD bioinformatics analyses were centralized at the Institut Curie. These analyses were performed within 48 h. Data were stored in a centralized database, along with the results of IHC performed in the Pathology Department. The bioinformatics’ platform provided a name-blinded technical report of the different molecular abnormalities identified (mutations, gene copy number alterations, and hormone receptors’ expression) to the Molecular Biology Board (MBB) of SHIVA. Automatic and manual controls generate query forms, and any change in the database was tracked. In addition, a seamless information system allowing the integration of the different types of clinical and molecular data was used. This information system ensured data management, data traceability, data analysis, query, and visualization. Being a prospective trial, SHIVA required different checkpoints at different levels from the biopsy to DNA extraction, quality control of the DNA and molecular data, as well as IHC, in order to ensure reproducibility of the results and a real-time follow-up. The molecular data were analyzed only when the checkpoints of the different steps were validated. For example, if the quality of the DNA was bad, the molecular platforms would not process the sample; if the quality of the NGS or CytoScan HD run was not acceptable than the bioinformatics pipeline would not process the data. In addition, only when all the clinical and molecular data were gathered, the bioinformatics’ analysis was performed and the report was edited (Fig. 9.2).
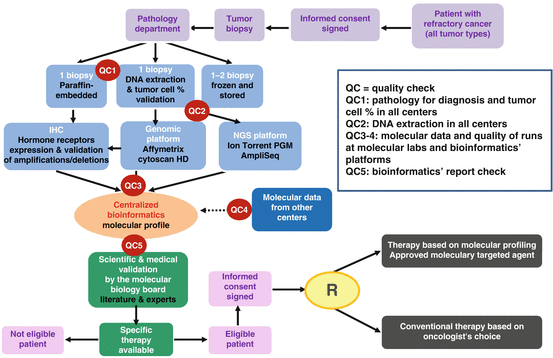
Fig. 9.2
SHIVA trial samples flow and quality checkpoints
One of the main important controls is to ensure the traceability of the patient’s samples and data. Thus, any additional quality control which can help in decreasing the sources of technical errors has to be implemented. Among them, a simple quality control can be set for all sequencing runs. The main idea is to be able to detect technical errors within a run, such as wrong patient identification, sample inversion, duplication, or contamination. One simple control which can be applied in practice is to select a list of highly polymorphic SNPs covered by the used sequencing panel. A set of 30 SNPs is usually enough to define a signature which is unique for each patient. This polymorphism signature can be then used to cluster the patient sequenced in the same run and detect any patient duplication. Another systematic control would be to check whether the somatic mutations detected, given one patient, are also detected in the other patients. This control can help detecting contamination or recurrent sequencing artifacts. It is therefore advised, as performed in frame of the SHIVA trial, to create an internal database with recurrent detected mutations which are likely to be false-positive events.
To achieve maximum clinical benefit, molecular results must be reported to clinicians in a clear and easily digestible way, yet with all supporting information necessary to interpret the significance of all molecular alterations that were detected. In this context, a detailed bioinformatics report was edited and provided anonymously to the MBB of the SHIVA trial. The report included few clinical information such as the sex of the patient, the histology type, and previous targeted therapies, necessary to the interpretation of the molecular results. In addition, the report provided a summary of the main alterations on the first pages on the report then a detailed picture of each gene mutational and DNA copy number alterations’ status (refer to Fig. 6.16).
9.2.4 Molecular Biology Board: A Treatment Algorithm
A major challenge in optimizing PM trials design is the appropriate use of relevant biomarkers to the MTA(s) in question and in case of several MTAs tested the setup of an appropriate algorithm. It is clear today that functional significance of some molecular alterations may differ across tumor types. Several molecular alterations are also observed in tumors, and few are considered “drivers.” The challenge is to be able to decipher the “driver” alterations and among these the alterations of major deleterious impact and those which are the determinants of inherent or acquired resistance in the context where very few alterations are validated in the clinics (refer to chapter 7). This issue is very tricky and complicates the setup of appropriate algorithms in PM trials. Based either on clinical or preclinical evidence, the functional significance of the biomarkers is continuously questioned not necessarily concerning its own function but most probably by the discovery of other alterations that can affect this function. Consequently, the presence of a multidisciplinary MBB is mandatory for all PM trials. The MBB of the SHIVA trial included biologists, physicians, bioinformaticians and the technical platforms’ managers, as well as basic and translational researchers. The MBB was in charge of the scientific validation and prioritization of the identified molecular abnormalities. Following the MBB, a final synthetic report was validated and signed by the accredited biologists. Previous therapy was taken into account by the physicians for the treatment recommendation. The whole process was set up in order to have less than 4 weeks elapsed between the biopsy and the day the MBB. The MBB got together on a weekly basis via a teleconference.
MTAs used in the experimental arm of the SHIVA trial were only drugs approved for clinical use in France. Single MTAs were selected following a predefined treatment algorithm (Le Tourneau et al. 2014), except for patients whose tumor harbored a mutation or an amplification of HER-2 who were proposed to be treated with trastuzumab and lapatinib given the overall survival benefit demonstrated in HER2-overexpressing metastatic breast cancer patients. The control arm was conventional chemotherapy as per oncologist’s choice. SHIVA treatment algorithm has been set up following several meetings with biologist and researchers and took into account the few alterations validated in the clinics and alterations described in the literature in a preclinical setting. In case of several alterations, the prioritization of the molecular alterations was discussed by the MBB based on specific criteria maintained throughout the trial to ensure reproducibility of treatment decision in all patients enrolled. For example, any molecular alteration (mutation, amplification, or deletion) was considered of a higher impact than hormone receptor expression. Only focal amplifications, defined as gene copy number ≥6 for diploid tumors and ≥7 for tetraploid tumors and an amplicon size of ≤10 Mb, are considered in the SHIVA algorithm. The amplifications ranging between 1 and 10 Mb require protein expression confirmed by IHC. Gene losses are defined as one copy for diploid tumors and one or two copies for tetraploid tumors, whereas gene deletions corresponded to zero copy. Losses of tumor suppressor genes such as PTEN required a confirmation by IHC of the loss of the protein. However, homozygous deletion did not require any further validation. Non-synonymous and nonrecurrent mutations were considered in the SHIVA algorithm. The MBB decides based on the literature whether or not these mutations are functionally relevant.
Related posts:
 Assessment of Biomarkers’ Predictive Value of Efficacy
Assessment of Biomarkers’ Predictive Value of Efficacy
 Introduction: Rationale for Precision Medicine Clinical Trials
Introduction: Rationale for Precision Medicine Clinical Trials
 The Key Role of Pathology in the Context of Precision Medicine Trials
The Key Role of Pathology in the Context of Precision Medicine Trials
 Microarrays-Based Molecular Profiling to Identify Genomic Alterations
Microarrays-Based Molecular Profiling to Identify Genomic Alterations
 Basis for Molecular Genetics in Cancer
Basis for Molecular Genetics in Cancer
 Bioinformatics for Precision Medicine in Oncology
Bioinformatics for Precision Medicine in Oncology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


