Example clinical reporting—MGMT
Negative result
RESULT: Gene Methylation NOT detected
Positive result
RESULT: Gene Methylation DETECTED
Methylation of the MGMT (0 (6)–methylguanine–DNA methyltransferase) gene was detected using methylation specific PCR technologies
Methylation score: 315.10
Unmethylated: <2.00
Methylated: >2.00
IDH1
Isocitrate dehydrogenase 1 and 2, soluble (IDH1/2) mutations are diagnostic and prognostic markers occurring early in gliomagenesis in the majority of low grade gliomas as well as secondary glioblastomas (GBM) [12]. The most common site of mutation in IDH1 is at codon 132 [13] and include R132H (most common), R132C, R132L, and R132G [12]. These mutations alter the function of the enzyme causing production of a potential oncometabolite, 2-hydroxyglutarate, instead of α-ketoglutarate. The presence of a mutated IDH1/2 gene compared to wild type IDH1/2 has been shown to be an independent predictor of better median overall survival in malignant and low grade gliomas (31 vs. 15 months for GBM, 65 vs 20 months for anaplastic astrocytomas and 110 vs 68 months in low grade gliomas) [14–16]. IDH2 mutations account for only 1–6 % of IDH mutations in gliomas but confer similar clinical benefit [17]. Reactive gliosis, pilocytic astrocytoma, and primary glioblastomas have wild type IDH; therefore, the presence or absence of IDH1/2 gene mutations can be a useful diagnostic tool for discerning WHO grade II gliomas from other lower grade/reactive lesions that may be histologically similar [18]. Additionally, IDH1/2 mutation status is important for distinguishing primary GBMs, which are characteristically wild type for IDH1, from secondary GBMs [12, 18]. There is currently insufficient evidence to support a predictive role for the IDH1/2 marker in gliomas; however, one study was able to show a better response to therapy in IDH1 mutated low grade gliomas treated with TMZ prior to anaplastic transformation [14].
The most common technique used for determining IDH1 mutation status is mutant specific immunohistochemistry (IHC) [18, 19]. IDH1 mutant specific antibodies have been shown to be 100 % sensitive and specific in detecting the R132H mutation [18, 20, 21]. Direct sequencing techniques may be more sensitive for detecting alternate mutations in IDH1 that are less common, though confer similar prognostic implications. DNA sequencing, however, is more labor intensive and requires specialized equipment not available at every center. Thus, IHC is more widely used in the clinical setting with the presence of a R132H mutation denoted by positive staining (Fig. 16.1).
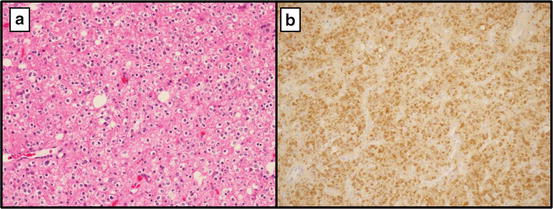
Fig. 16.1
(a) Mixed oligoastrocytoma, WHO grade II at 20× showing a predominance of oligodendroglial-like cells (b) R132H mutant specific IHC positive
1p19q
The codeletion that occurs as a result of an unbalanced translocation of 19q and 1p is considered a hallmark of most oligodendroglial tumors. The nearly absolute specificity of the 1p19q codeletion in oligodendrogliomas makes this marker a useful diagnostic tool for distinguishing these neoplasms from other histologically similar lesions such as central neurocytomas, clear cell ependymomas, and dysembryoplastic neuroepithelial tumors (DNET) [22–24]. The presence of this codeletion is also helpful in differentiating anaplastic oligodendrogliomas from the small cell variant of glioblastoma [25]. The 1p19q status cannot be considered in the classical sense to be predictive of response to a specific therapy; however, patients diagnosed with oligodendroglial neoplasms harboring this genetic alteration have significantly better outcomes following treatment compared to those with non-deleted tumors [26, 27]. Patients treated for anaplastic oligodendrogliomas (WHO grade III) with the 1p19q codeletion were found to have significantly longer median survivals, 7 vs. 2.8 years [28, 29]. In the absence of chemotherapy and radiation, both intact and 1p19q codeleted lesions exhibit similar natural histories suggesting that a defect associated with this alteration manifests only in the presence of adjuvant therapy [30]. Although not predictive for response to specific therapies, 1p19q mutational status can impact the timing of treatment as some neuro-oncologists will initially withhold radiotherapy in 1p19q codeleted patients to avoid potential long term toxicity and cognitive decline when significantly longer overall survival is expected [31].
The 1p19q mutational status in gliomas can be determined using several techniques including PCR-based microsatellite loss of heterozygosity (LOH), single nucleotide polymorphism analysis, and fluorescent in situ hybridization (FISH) [32]. FISH is the most common method used in the clinical setting because of reliability in detecting the codeletion, ease of use with FFPE tissue, and the ability to discern areas of neoplastic tissue in a heterogeneous sample to reduce dilution effect by nonneoplastic tissue. This morphologic basis makes FISH testing particularly attractive to pathologists for 1p19q analysis in glial neoplasms. FFPE sections of brain tumors suspected to be of oligodendroglial origin are separately hybridized with specific test and control probes for chromosomes 1 and 19. For chromosome 1, the fluorescent test probe is typically localized to 1p36 and the control probe localized to 1q25. Test and control probes for chromosome 19 are localized to 19q13 and 19p13, respectively. A signal ratio of test to control probes for a clinically codeleted neoplasm is < 0.8. A ratio of 0.9 to 1.05 is considered intact [33] (Fig. 16.2).
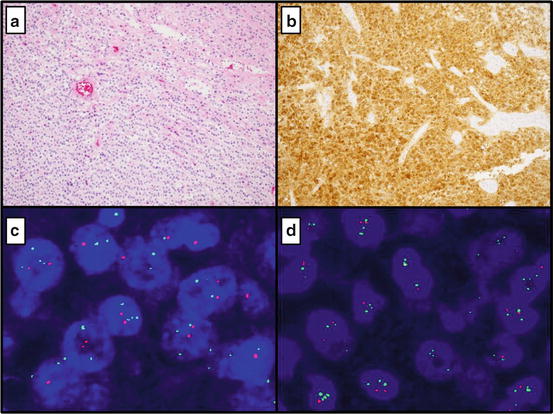
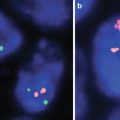
Fig. 16.2
(a) Oligodendroglioma at 10× showing a uniform population of cells with prominent perinuclear halos and thin vasculature (b) Mutant specific (R132H) immunohistochemistry with diffuse positivity (c) FISH for 1p deletion (Average number of 1p36 signals per cell:1.65, Average number of chromosome 1 control signals per cell: 2.80; positive for 1p deletion) (d) FISH for 19q deletion (Average number of 19q13 signals per cell: 1.69, Average number of chromosome 19 control signals per cell: 2.91; positive for 19q deletion)
Example clinical reporting—1p19q | |
Negative result | Karyotype : nuc ish(TP73,ANGPTL)x1 ~ 2[100], (ZNF44,GLTSCR2)x1 ~ 2[100] FISH results: Negative for a deletion of 1p36 (ratio: 1.09) and negative for a deletion of 19q13 (ratio: 1.01) |
Positive result | Karyotype : nuc ish 1p36(TP73x1,ANGPTLx2)[92/100] FISH results: POSITIVE for a deletion of 1p36 (ratio: 0.597) nuc ish 19p13(ZNF44x2,GLTSCRx1)[87/100] FISH results: POSITIVE for a deletion of 19q13 (ratio: 0.598) |
EGFR
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that when activated, induces intracellular signaling that promotes cell division, migration and inhibition of apoptosis [34]. Amplification of this gene hypersensitizes tumor cells to extracellular EGF, leading to a survival advantage over non-amplified neoplasms. EGFR is abnormally activated in 70 % of solid tumors including glioblastomas [15, 35]. EGFR amplification is a hallmark of high-grade gliomas and is considered a diagnostic marker for distinguishing small cell variant glioblastomas from anaplastic oligodendrogliomas [25, 36]. This marker is also useful for distinguishing the more common primary GBMs from secondary GBMs. EGFR amplification is nearly mutually exclusive to IDH1/2 mutations and 1p19q codeletions [25, 37–40]. The prognostic and predictive value of EGFR amplification in gliomas is not clearly defined [41–46]. In other types of neoplasms such as non-small-cell lung cancer (NSCLC), specific EGFR mutations have been shown to be predictive of response to small molecule EGFR tyrosine kinase inhibitors (gefitinib) [47–49]. Phase II trials using gefitinib to treat recurrent GBM patients with EGFR alterations showed little effect on event free survival [50]. The discrepancy in efficacy of tyrosine kinase inhibitors in GBMs and lung cancers may be due to poor penetration of the blood brain barrier (BBB), interactions with antiepileptics commonly used in brain tumor patients, or innate differences in the function of EGFR in GBM vs. NSCLC [51, 52]. For example, 50 % of EGFR amplified GBMs have a constitutively active truncated mutant variant of the protein (EGFR vIII) [25, 42, 53]. This variant (EGFR vIII) may be inherently resistant to gefitinib [51].
Alterations in EGFR can be evaluated using various methods including FISH, mutant specific IHC and direct sequencing techniques. For FISH analysis, amplification of EGFR is detected using a test probe to EGFR and a control probe to chromosome 7 centromere (CEP7). EGFR amplified tumors require a ratio of >2:1; however, these tumors typically have a test to control probe ratio greater than 10 [32, 54]. Commercially available FISH probes do not distinguish the truncated mutant variant (EGFRvIII) from wild type [42]; however, amplification is demonstrated in these cases nonetheless. The presence of this mutant variant can be determined using direct sequencing techniques and more commonly by mutant specific IHC [25, 41, 55]. Overexpression of EGFR detected using IHC with wild type (WT) antibodies does not correlate with amplification of the EGFR gene as determined by FISH. Additionally, WT EGFR expression by IHC can be seen in oligodendrogliomas of all grades negating the potential for diagnostic utility with respect to distinguishing small cell GBM from anaplastic oligodendroglioma [56–58] (Fig. 16.3).
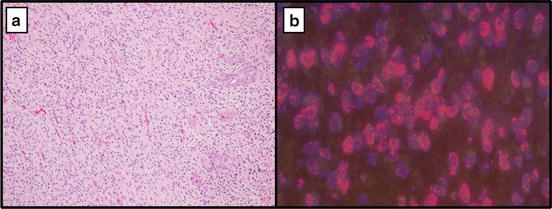
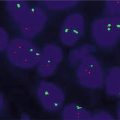
Fig. 16.3
(a) Glioblastoma at 10× showing hypercellularity, atypia, and endothelial proliferation. (b) FISH for EGFR amplification (Average number of EGFR signals per cell: 35.4, Average number of control signals per cell: 1.85; Positive for amplification of EGFR (ratio: 19.1))
Example clinical reporting—EGFR | |
Negative result | Karyotype: nuc ish(EGFRx1 ~ 3,D7Z1x1 ~ 3)[107] Average number of EGFR signals per cell: 1.79 Average number of control signals per cell: 1.79 FISH results: Negative for amplification of EGFR (ratio: 1.0) |
Positive result | Karyotype: nuc ish (EGFRx10 ~ 50,D7Z1x1 ~ 3)[60] Average number of EGFR signals per cell: 26.68 Average number of control signals per cell: 1.96 FISH results: POSITIVE for amplification of EGFR (ratio: 13.6) |
Pediatric CNS Tumors
Gliomas
KIAA1549-BRAF Gene Fusion
Pilocytic astrocytoma (PA) is the most common pediatric brain tumor and the most frequent childhood glioma. The most classic histologic appearance of PA is that of a biphasic astrocytic neoplasm with alternating compact and loose arrangements of cells with elongated “piloid” processes admixed with Rosenthal fibers and eosinophilic granular bodies. Unfortunately, PA is well known for its diagnostically problematic ability to mimic a wide variety of other neoplasms, including high grade infiltrative gliomas and others with significantly different biologic potential, prognosis, and applicable treatment options (Fig. 16.4).
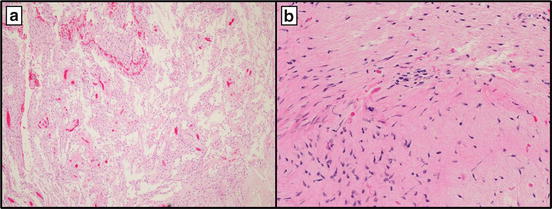
Fig. 16.4
(a) Pilocytic astrocytoma at 10× showing loose arrangements of cells with elongated “piloid” processes and (b) compact/dense areas with abundant Rosenthal fibers at 40×
Though most are sporadic, there is a well-established association of PA and Neurofibromatosis Type 1 (NF1) with germline mutations of the NF1 gene [59]. The resultant loss of the NF1 gene product neurofibromin leads to activation of the mitogen activated protein kinase (MAPK) signaling pathway. More recently, a number of groups have identified a low-level gain at chromosome 7q34 resulting in a tandem duplication of the BRAF gene in most sporadic PAs [60, 61]. This duplication results in a KIAA1549-BRAF fusion that is nearly specific to PAs and absent or exceedingly rare in other low grade or high grade infiltrative astrocytomas [62–65]. KIAA1549-BRAF fusion is more frequent in PAs arising in the cerebellum (up to 80 %) compared to those arising in the brainstem, and hypothalamic/optic pathway (approximately 60 %) or the cerebral hemispheres (14–19 %) [62, 65–67]. KIAA1549-BRAF fusion detection is particularly helpful in differentiating those uncommon cerebellar PAs with a diffuse pattern of growth (so-called “diffuse variant”) from WHO grade II diffuse astrocytomas in this region [68]. BRAF fusion detection also may be helpful in distinguishing PAs with atypical radiographic and/or histologic features from pleomorphic xanthoastrocytoma (PXA) or malignant glioma [69]. KIAA1549-BRAF fusion therefore represents a diagnostically useful molecular marker for differentiating PA from potential diagnostic mimics [70, 71]. The prognostic or therapeutic predictive utility of KIAA1549-BRAF fusion is not yet established, though a number of clinical trials for pediatric PAs utilizing drugs targeting the MAPK pathway are ongoing [67, 72]. It should be noted that in contrast to pediatric PAs, KIAA1549-BRAF fusion is less commonly detected in PAs arising in the adult population [73] and has been encountered in occasional adult diffuse gliomas, particularly oligodendrogliomas, often together with IDH mutations and 1p/19q codeletions [74].
A number of approaches have been employed in the clinical testing of BRAF fusions. RT-PCR may be used to directly amplify the fusion transcript. Though this procedure is quite specific, assessment is limited to predetermined regions and requires sufficient RNA material from tumor samples [62, 63, 72]. Pyrosequencing methods for detecting BRAF copy number gains compared favorably with RT-PCR methodology and can provide a sensitive, cost-effective alternative to RT-PCR [75]. Fresh or FFPE tissue samples may be used for either of these techniques; however, FISH is at present the best method for testing of the KIAA1549-BRAF fusion in FFPE. Detection of BRAF duplication can be accomplished by determining the ratio of signals of a fluorescent probe that includes the centromeric end of BRAF versus a ploidy control probe (i.e., CEP7); a ratio of test: control probe ≥ 1.15 and > 20 % of cells with relative BRAF gain would be considered positive. This method is considered indirect evidence of the KIAA1549-BRAF fusion and would typically be visualized as nuclei containing 2 CEP7 control signals, 2 BRAF signals, and a third smaller BRAF signal adjacent to one of the larger BRAF signals [67]. An additional FISH method employs separately labeled probes targeting BRAF and KIAA1549 genes, typically tagged red and green respectively. The presence of nuclei with one fused red/green signal and a normal pair of red and green signals is considered positive for a KIAA1549-BRAF fusion [76–78]. Both FISH methods allow for direct visualization of BRAF alterations in tumor cells and can be used on small tumor samples. The indirect FISH technique is more difficult to interpret, particularly in FFPE tissue samples in which truncation of small signals could result in false negatives and more interobserver variability.
BRAF V600E Mutations
In addition to KIAA1549-BRAF fusions, activating mutations of BRAF have been detected in a variety of pediatric glial tumors. The most common alteration is the V600E mutation (BRAF V600E) caused by a point mutation at codon 600 which results in a substitution of glutamic acid for valine. This mutation has been described in a wide variety of tumors outside of the CNS and is found most frequently in melanoma, lung, and colon cancers [79]. Although BRAF V600E mutations may be found in a small subset of PAs, particularly in extra-cerebellar examples, they are exceedingly more common in other pediatric low grade gliomas [80]. Up to 75 % of pleomorphic xanthoastrocytomas (PXA) have a BRAF V600E mutation, including those with anaplastic features [80–83]. Nearly all temporal lobe PXAs harbor this mutation compared with 50 % of non-temporal examples [84]. Similarly, this alteration is present in up to 40 % of gangliogliomas and subependymal astrocytomas as well as a significant proportion of dysembryoplastic neuroepithelial tumors (DNET) [81, 82].
Despite its significant histologic pleomorphism, PXA is less biologically aggressive than pleomorphic infiltrative gliomas, thus earning it a WHO grade II designation. From a clinical perspective, it is imperative to differentiate PXA from other gliomas as treatment and prognosis are significantly different. Demonstration of the BRAF V600E mutation serves as a useful diagnostic marker in this setting. Although BRAF V600E mutation may be encountered in either PA or PXA, detection of KIAA1549-BRAF fusion would confirm the diagnosis of PA as this alteration is not present in PXA. When glioblastoma is in the differential diagnosis, the presence of a BRAF V600E mutation would strongly favor PXA as this mutation is rare in GBM [80, 81, 85, 86]. Combined BRAF V600E and cyclin-dependent kinase inhibitor 2A (CDKN2A also known as P16) loss may be detected in up to 50 % of PXAs, but this signature is not observed in rhabdoid or giant cell glioblastomas, both potential histologic mimics [84, 86]. Unfortunately, BRAF V600E is not useful in separating PXA from epithelioid GBM as 50 % of the later may have this mutation [86]. Additionally, recent studies have indicated that pediatric GBMs have a slightly higher rate of BRAF V600E compared to GBMs in adults (12 versus 8 %) [87–89] (Fig. 16.5).
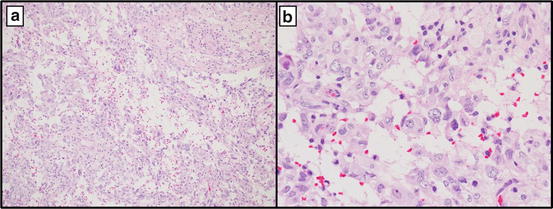
Fig. 16.5
Pleomorphic xanthoastrocytoma at 10× (a) and 40× (b) showing a hypercellular lesion with atypical cells and scattered xanthomatous cells (b)
The prognostic utility of BRAF V600E mutational analysis is uncertain [82]. In one study, BRAF V600E PXAs had a significantly longer overall survival compared with those lacking the mutation [83]. In contradistinction, others have found BRAF V600E mutation to be associated with an increased risk of malignant progression in pediatric low grade gliomas [89, 90]. Additionally, there is some evidence that suggests BRAF V600E may represent an important biomarker of targeted therapy response. Recent reports have shown that treatment of BRAF-mutated anaplastic PXA and recurrent GBM with vemurafenib, a BRAF inhibitor, may be effective [91, 92]. However, additional prospective studies/clinical trials are necessary to determine whether BRAF mutational analysis will serve a key clinical prognostic and/or predictive role in the workup of these glial tumors.
The gold standard for BRAF mutation analysis is direct sequencing; however, this technique is complex, labor-intensive, and requires specialized equipment not universally available in all clinical laboratories. Development of BRAFV600E-mutant specific monoclonal antibodies has provided a cost-effective alternative to sequencing techniques for routine clinical testing [93, 94]. The VE1 clone of the BRAFV600E-mutant specific antibody provides strong cytoplasmic staining with 100 % specificity and sensitivity versus standard molecular methods for detecting BRAFV600E mutation [93]. This IHC method is able to accurately demonstrate BRAFV600E in a variety of tumor types including gliomas and is amenable to very small biopsy samples, which might otherwise be inadequate for sequencing analysis. Interpretation is straightforward, though careful validation is important and care should be taken to not over interpret weak nonspecific staining.
H3F3A/ATRX/DAXX Mutations
Although glioblastoma (GBM) represents the most common primary malignant CNS tumor in adults, it is uncommon in children. Pediatric GBMs share a similar histologic appearance and aggressive biologic behavior as their adult counterparts; however, their anatomic distribution is notably different. In addition to the typical cerebral locations of adult GBMs, pediatric high grade gliomas may also arise within deep grey structures as well as the brainstem in the form of diffuse intrinsic pontine glioma (DIPG). Pediatric GBMs harbor a significantly different set of genetic alterations than those of adult tumors with minimal overlap. Specifically, mutations in the H3 histone family 3A (H3F3A)–α-thalassemia/mental retardation syndrome X-linked (ATRX)–death-domain associated protein (DAXX) chromatin remodeling pathway are present in 44 % of pediatric GBM [95, 96]. The H3 histone family 3A (H3F3A) gene mutation leads to an amino acid substitution at either K27 or G34. These mutations are present in up to 60 % of DIPGs and 33 % of non-brainstem pediatric GBMs [95]. Similarly, mutations in histone cluster 1, H3b (HIST1H3B), an isoform of H3F3A, have also been documented in nearly 20 % of DIPGs [97]. Thus, approximately 80 % of DIPGs will harbor some form of H3-related mutation [97]. Interestingly, the K27 and G34 mutations correspond to distinct pediatric GBM subgroups, with the majority of K27 mutations found in DIPGs and deep midline tumors and a mix of K27 and G34 mutations found in non-brainstem pediatric GBMs [96–98]. Mutations involving ATRX (α-thalassemia/mental retardation syndrome X-linked) and DAXX (death-domain associated protein), both gene products involved in the chromatin remodeling complex requisite for H3F3A incorporation at telomeres, have been identified in 31 % of pediatric GBMs as well as in all tumors harboring H3F3A G34 mutations [95].
From a diagnostic standpoint, H3F3A mutations have been found to be specific to GBMs arising in children and young adults and have not been found in other CNS tumors. A variety of other pediatric CNS tumors may pose a significant diagnostic challenge given the multitude of histologic appearances noted in pediatric GBMs. These include PXA, PA, medulloblastoma, primitive neuroectodermal tumor (PNET) and AT/RT. Demonstration of H3F3A (or ATRX or DAXX) mutations may assist in securing an accurate tumor classification and allow for appropriate treatment. Gene sequencing techniques are the gold standard of testing for these mutations. However, IHC is much more cost-effective and more suitable for clinical detection. Unfortunately, a commercially available mutant specific antibody has yet to be developed. A single large multi-institutional study utilized antibodies against oligodendrocyte lineage transcription factor 2 (OLIG2), forkhead box G1 (FOXG1), and IDH1R132H to classify these tumors into specific mutation defined GBM subgroups. In this study, there was excellent correlation between sequencing-derived mutation status and the IHC subgroups with H3F3A K27 corresponding to OLIG2+/FOXG1−, and H3F3A G34 corresponding to OLIG2−/FOXG1+ staining [96]. Given an observed difference in overall survival for these three mutation-defined subgroups (K27 associated with shortened survival, versus prolonged survival with G34 and particularly with IDH1 R132H) [98, 99], assessment for these IHC markers may well become routine practice in the workup of pediatric GBM.
Ependymomas
Ependymomas may occur in any age group and arise throughout the brain and spinal cord. However, they predominate in children and represent the third most common pediatric CNS tumor [100]. Pediatric ependymomas most commonly arise within the posterior fossa and, though they generally carry a poor prognosis, there remains significant variability in patient outcomes. Extent of tumor resection, patient age, and histologic grade (WHO grade II versus III) have been found to represent important prognostic variables [101, 102]. More recently, two clinically and molecularly distinct subtypes of pediatric posterior fossa ependymomas were identified by multiple independent genome-wide profiling studies [103–105]. Group A tumors show a mesenchymal profile together with infrequent chromosome 1q gains as well as younger age of occurrence (median age less than 3 years), anaplastic (WHO grade III) features, higher incidence of metastasis, significantly shorter progression-free and overall survival compared with Group B tumors. Group B tumors tend to occur in young adults and adolescents and harbor numerous cytogenetic abnormalities including gains of chromosomes 9q, 15q, and 18q and losses involving chromosomes 6q and 22q [103, 104].
The ability to differentiate between Group A and Group B posterior fossa ependymomas provides neuro-oncologists with a more accurate prognostic assessment of the biologic potential of individual posterior fossa ependymomas. The most practical testing for ependymoma group determination is with immunohistochemical methods. A large series of ependymomas found that laminin alpha-2 (LAMA2) and neural epidermal growth factor-like-2 (NELL2) represent excellent markers for Group A and Group B tumors, respectively [78, 103]. A more comprehensive analysis might include additional testing by FISH or array comparative genomic hybridization (CGH) for copy number alterations to demonstrate chromosome 1q gain in Group A tumors or gains of 9q, 15q, 18q or loss of 6q or 22q in group B tumors (Fig. 16.6).
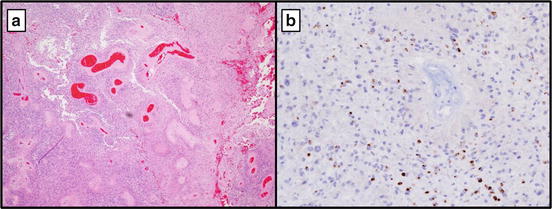
Fig. 16.6
Ependymoma, WHO grade II at 10× showing perivascular pseudo-rosettes (a). (b) Epithelial membrane antigen (EMA) with typical perinuclear dot-like pattern by IHC
Embryonal Tumors
Medulloblastoma Molecular Subgroups
Medulloblastoma represents the most common childhood malignant CNS tumor. Traditionally their classification has been based entirely on histopathologic features with classic, large cell, anaplastic, desmoplastic nodular, and extensively nodular as the accepted recognized variants [100]. Unfortunately, this classification alone does not provide accurate prognostic information as there is still significant variability of outcomes in these histologic groups. Genome-wide molecular characterization studies have revealed four molecular subgroups of medulloblastomas each with their own molecular signature as well as prognostic and treatment implications [78, 106].
WNT Group
The WNT group is the best characterized of the subgroups, though it accounts for the smallest proportion of medulloblastomas overall (approximately 10 %) [107]. Germline mutations of adenomatous polyposis coli (APC), a wingless-type MMTV integration site family (WNT) pathway inhibitor, predispose patients with Turcot syndrome to medulloblastomas. Conversely, sporadic medulloblastomas of this group bear catenin (cadherin-associated protein) beta 1 (CTNNB1) somatic mutations. WNT medulloblastomas are rare in infants, have classic histology with rare anaplastic examples, and are characterized by nuclear β-catenin staining by immunohistochemistry. Monosomy 6 and dickkopf WNT signaling pathway inhibitor 1 (DKK1) positive immunostaining are also typically present. Of the four subgroups, WNT medulloblastomas have the best prognosis with uncommon metastasis and prolonged survival [106–108]. These patients may also be more amendable to treatment de-escalation, though this option needs further investigation.
SHH Group
Medulloblastomas in the Sonic Hedgehog (SHH) signaling pathway subgroup have demonstrable mutations of patched 1 (PTCH1), suppressor of fused homolog (SUFU), smoothened frizzled class receptor (SMO), or amplification of GLI family zinc finger 1 or 2 (GLI1/GLI2) (2120). Germline PTCH1 mutations are also associated with the development of medulloblastomas in the context of Gorlin syndrome. SHH medulloblastomas are most common in infants and adults, but are rare in children. The vast majority of nodular desmoplastic medulloblastomas belong to this group, although approximately half of the SHH subgroup are of some other histology. SHH medulloblastomas can be accurately identified by immunopositivity for secreted fizzles-related protein (SFRP1) or GRB2-associated binding protein 1 (GAB1), as well as deletion of chromosome 9p detectable by FISH [106–108]. Importantly, a recent large multinational study determined that SHH medulloblastomas can be further stratified into three prognostically significant risk groups: GLI2 amplification alone or 14q loss combined with M+ (positive for metastatic disease) status correlates with a high risk group, whereas those lacking all of these markers constitute a low risk group having a prognosis similar to the WNT group. The remainder fall into a standard risk group with intermediate prognosis similar to group 4 [109] (Fig. 16.7).
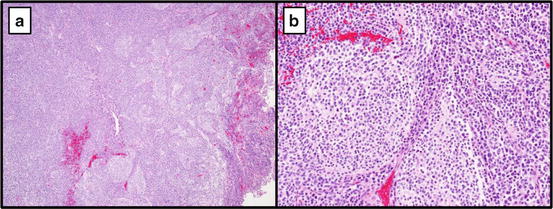
Fig. 16.7
Medulloblastoma, nodular desmoplastic type 10×(a) and 40× (b). Most likely to belong to the SHH molecular subgroup
Group 3
The molecular alterations and pathways involved in subgroup 3 are less well characterized than the WNT or SHH subgroups. Group 3 medulloblastomas are more common in male infants and children and are exceedingly rare in adults. Although many have a classic histology, the majority of anaplastic/large cell medulloblastomas cluster in this subgroup often with metastatic disease at presentation. Originally classified based upon a specific transcriptional profile, accurate categorization can be accomplished through demonstration of immunopositivity for natriuretic peptide receptor C (NPR3) [106–108]. Like SHH group tumors, Group 3 medulloblastomas can be further subclassified into prognostically significant risk groups. Tumors with detectable v-myc avian myelocytomatosis viral oncogene homolog (MYC) amplification, Iso17q [78], or evidence of metastatic disease at presentation are considered high risk, with frequent tumor recurrence and short survival. In contradistinction, tumors lacking any of those three features are considered standard risk with prognoses similar to group 4 tumors [106, 109, 110] (Fig. 16.8).
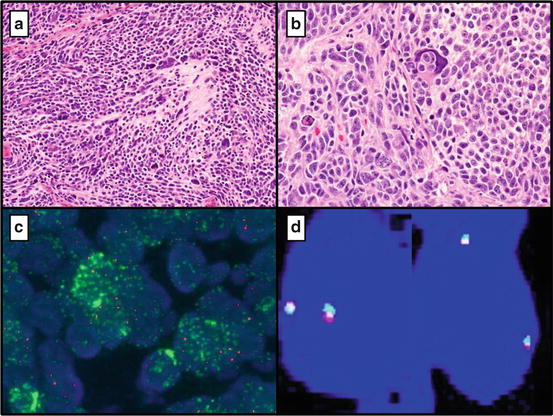
Fig. 16.8
(a) Anaplastic Large Cell Medulloblastoma (Group 3) (10×) and 40× (b). (c) Positive MYCN (N-MYC) amplification by FISH (The mean number of signals/cell for the test probe is 21.2, with a mean of 3.10 signals for the control probe.) (d) Negative for MYC fusion by FISH (Break apart probe intact)
Group 4
Like Group 3 tumors, Group 4 medulloblastomas were originally defined by their unique transcription profile. Most are of classic histology arising preferentially in children with a male predominance. Detection of potassium voltage-gated channel, shaker-related subfamily member 1 (KCNA1) by immunohistochemistry can reliably classify Group 4 medulloblastomas [106–108]. Group 4 tumors can be further stratified into prognostically significant risk groups based on additional molecular testing and clinical parameters. The medulloblastomas of this group with demonstrable whole chromosome 11 loss (Chr11 loss) or chromosome 17 gain (Chr17 gain) fall into a low risk stratum with excellent prognosis and prolonged survival even in the presence of aggressive histology (anaplastic/large cell) or M+ disease. Conversely, those Group 4 patients presenting with metastases but without these chromosomal abnormalities would otherwise be considered as high risk. The remainder fall into a standard risk group with intermediate prognosis similar to SHH [109].
Determination of subgroup affiliation together with selective cytogenetic workup for risk stratification will provide for accurate determination of individual patient prognosis and risk-directed treatment options useful to neuro-oncologists. Accurate medulloblastoma subgroup determination can be made via immunohistochemistry, with positivity for Beta-catenin and DKK1 in the WNT group, SFRP1 in the SHH group, NPR3 in Group 3, and KCNA1 in the Group 4 tumors [107, 108]. Further risk-stratification in SHH, Group 3, and Group 4 tumors can be accomplished by employing interphase FISH assays using the following cytogenetic biomarkers: GLI2 and 14q in SHH group tumors, MYC, 17p, and 17q in Group 3 tumors, and chromosome 11, 17p, and 17q in Group 4 tumors [109]. 17p and 17q FISH probes are paired together for analysis of SHH and Group 3 tumors; nuclei with iso17q would contain three 17q signals with two 17p signals, whereas Chr17 loss would show loss of single 17p and 17q signals. Alternately, these cytogenetic copy number alterations could be accurately assessed in a single array CGH assay; however, this testing platform is fairly cost prohibitive for widespread clinical implementation.
SMARCB1 (INI1)
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare type of CNS embryonal tumor that exhibits extremely aggressive behavior and early metastases [100]. It most frequently arises in children under 5 years of age and is resistant to conventional medulloblastoma therapies [111, 112]. Accurate diagnosis of AT/RT from other CNS tumors is paramount in appropriately directing treatment options. Unfortunately, AT/RT is well known for its ability to display a wide array of histomorphologic features, and the diagnostic “rhabdoid cells” with eosinophilic cytoplasmic filamentous inclusions may be difficult to find. When embryonal small round blue cells predominate, these tumors may be easily confused with medulloblastoma or CNS PNET. Adding to the potential diagnostic dilemma, meningioma and glioblastoma can display a rhabdoid phenotype. The vast majority of AT/RTs have been found to harbor alterations of chromosome 22q11.2 with the SMARCB1 (also known as INI1, BAF47, hSNF5) gene being mapped to this region. Deletion and/or mutation of SMARCB1 has been detected in the majority of AT/RTs (up to 75 %) having demonstrable monosomy 22 or 22q deletion [111–113]; whereas these alterations are generally not detected in PNETs and medulloblastoma [114–116].
The presence of an SMARCB1 alteration can be assessed by a variety of molecular techniques. FISH analysis has predominately relied upon “laboratory developed” probes targeting the SMARCB1 locus and thus, testing has only been available at a few specialized centers [117]. This type of FISH analysis provides a rapid assessment for SMARCB1 deletions on FFPE samples; however, up to 30 % of AT/RTs have no detectable deletions necessitating SMARCB1 mutational analysis. Immunohistochemical staining for the SMARCB1 (INI1/BAF47) protein has demonstrated a universal loss of nuclear expression for SMARCB1 in AT/RT with excellent correlation between this immunohistochemical assay and other molecular techniques [118, 119]. Nuclear staining with anti-BAF47 likewise accurately differentiates rhabdoid variants of glioblastoma and meningioma from AT/RT [120]. Therefore, this loss of nuclear expression of SMARCB1 identified by IHC is considered the diagnostic hallmark of AT/RT, which can be easily incorporated into most clinical laboratory settings.
Dural Based Tumors
Meningioma
1p and 14 q Deletions
Malignant progression in meningiomas appears to be associated with the sequential accumulation of various genetic abnormalities with common alterations in chromosomes 1p and 14q [121–132]. Allelic losses on chromosome 1p have been shown to be of prognostic significance with alterations being more common in higher grade meningiomas. Additionally, 1p deletions are also associated with increased risk of recurrence/progression, although the significance of this finding is less clear when results are adjusted for histologic grade [121–137]. Likewise, loss of chromosome 14q is associated with poor prognostic features such as higher grade lesions and increased risk of recurrence. Three year survivals for patients diagnosed with meningiomas with 14q deletions compared to those with wild type lesions were 20.5 % vs 68.6 % respectively [121]. 14q deletions, when present in grade I meningiomas, are also associated with a higher risk of recurrence, 17 % vs 50 % recurrence in deleted tumors. 1p and 14q deletions used along with chromosome 22q loss, which is a well-known frequent occurrence in meningiomas, can be diagnostically useful for differentiating meningiomas from other dural-based tumors such as hemangiopericytoma, metastatic carcinoma, superficial glioblastoma and gliosarcoma [123, 125, 138–143]. There is currently no known predictive value for these markers.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


