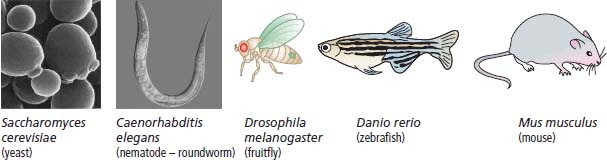
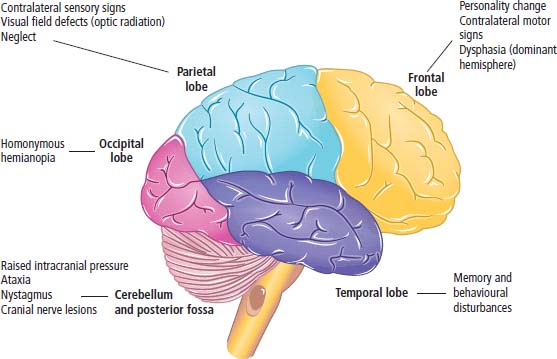
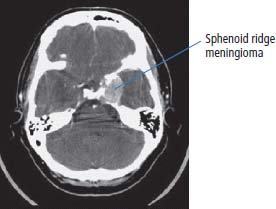
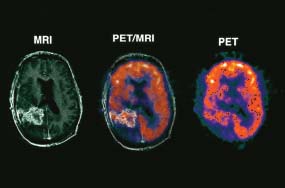
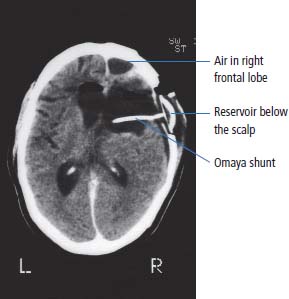
6 The first recognized resection of a primary brain tumour was performed in 1884 by Rickman Godlee in collaboration with the Westminster Hospital neurologist Alexander Bennett. It should be remembered that the removal of the cerebral cortex tumour was performed before any diagnostic imaging was available (X-rays were discovered in 1895 by Wilhelm Röntgen), but even then the surgeon knew to operate on the contralateral side to the clinical signs! Scientists use animal models to study genetics, development and oncogenesis, and the most common models are mice (see onco-mice in Box 1.1), fruit flies (Drosophila melanogaster) and nematode worms (Caenorhabditis elegans) (Figure 6.1). However, fish have also turned out to be useful subjects – in particular zebrafish (Danio rerio), which are tropical freshwater minnows with five horizontal stripes running from their mouth to the caudal fin. Zebrafish are the vertebrate model for studying the genetics of embryonic development because the embryos are transparent and the genome sequence is known and can readily be modified by knockouts. A less well-known fish model of cancer is the damselfish (Pomacentrus partitus) that lives on coral reefs and has several vertical stripes. Deb and Flo in Finding Nemo are damselfish, so here is an excuse to watch that masterpiece of subtlety and metaphor again. Damselfish neurofibromatosis is a naturally occurring, neoplastic disease of these fish that consists of multiple neurofibromata and neurofibrosarcomas. It has been proposed as an animal model for neurofibromatosis type 1 in humans. However, whilst von Recklinghausen’s neurofibromatosis type 1 is vertically transmitted as an autosomal dominant trait, damselfish neurofibromatosis is transmitted horizontally between fish and is thought to be due to an oncogenic retrovirus named damselfish neurofibromatosis virus (DNFV). There were 9365 people diagnosed with a brain tumour in 2011 in the United Kingdom and 4975 deaths from brain tumours. Brain tumours are slightly more common in men than in women. Fifteen per cent of adults diagnosed with brain tumours survive for 5 years. A further 400 children are diagnosed with central nervous system (CNS) tumours in the United Kingdom each year. CNS tumours are the second most common cancer in childhood after leukaemia and account for 27% of all cancers in children. Metastases to the brain are about 10 times more common than primary brain tumours. The most common primary tumour sites amongst patients with brain metastases are lung, breast, melanoma and kidney. In addition, nasopharyngeal cancers may directly extend through the skull foramina. Meningeal metastases occur with leukaemia and lymphoma, breast and small-cell lung cancers, and from medulloblastoma and ependymal glioma as a route of spread. Primary tumours of the CNS account for 1.5% of all cancers and 3% of cancer deaths (Table 6.1). About 58% of CNS tumours arise in the brain, 23% in the meninges, 11% in the pituitary and 8% in the spinal cord. Similarly, astrocytomas account for 34% of CNS tumours, meningiomas for 21% and pituitary adenomas for 8%. The World Health Organization (WHO) International Classification of Diseases (ICD) attributes a code to all diseases and the current version is ICD-10, the first version of the ICD that has removed homosexuality from its ridiculous classification as a mental illness! CNS tumours are broadly grouped into malignant or benign tumours and the grade of the tumours in the WHO classification can be loosely translated into aggressive (generally grades II–IV) and indolent (generally grade I) tumours. Most (95%) astrocytomas are aggressive, but only 8% of meningiomas and 1–2% pituitary tumours are aggressive. Figure 6.1 The oncologist’s menagerie. The five most common animal models used in laboratory studies of cancer. Although the cause of most adult brain tumours is not established, a number of inherited phakomatoses are associated with brain tumours. Phakomatoses are a group of familial conditions with unique cutaneous and neurological manifestations and dysplasias of a number of organ systems. Phakomatoses include: The first three of these are associated with brain tumours; von Recklinghausen’s neurofibromatosis with cranial and root schwannomas, meningiomas, ependymomas and optic gliomas (see Figure 2.15); tuberous sclerosis with gliomas and ependymomas; and von Hippel–Lindau disease with cerebellar and retinal haemangioblastoma (Table 6.2). In addition, an increased incidence of brain tumours is a feature of Gorlin’s basal nevus syndrome (medulloblastoma), Turcot syndrome (gliomas) and Li–Fraumeni syndrome (glioma) (see Table 2.3). Table 6.1 UK registrations for primary brain cancer 2010 Table 6.2 Phakomatoses associated with brain tumours High-dose ionizing radiation to the head region administered in the past for benign conditions such as scalp tinea capitis fungal infection (ringworm) increases the risk of nerve sheath tumours, gliomas and meningiomas. There is much public concern that low-frequency non-ionizing electromagnetic fields such as those emitted by 60 Hz power cables may increase the risk of brain tumours, but there is no consistent evidence to support this hypothesis. Similarly, despite scares, there is no evidence to support an association with wireless radiofrequency devices such as mobile phones. This joins the long list of things that cause cancer according to the Daily Mail. It includes Facebook, deodorant, hair dye, talcum powder, mouthwash, tooth whitener, oral sex, chips and chocolate. Primary nervous system tumours may be glial tumours, non-glial tumours or primary cerebral non-Hodgkin’s lymphoma (Table 6.3). As you may recall from embryology lectures, the early embryo has three distinct germ layers of cells: Neuroectodermal tumours are classified on the basis of the predominant cell type and include tumours of both central and peripheral nervous system derived cells. After embryonic development ceases, neurons do not divide, but glial cells retain the ability to proliferate throughout life and thus most adult neurological tumours are derived from glial cells and are named gliomas. Glial cells (from the Greek term for glue) surround the neurons holding them in place, supplying them with nutrients and insulating them from one another; glia also remove pathogens and dead neurons. Table 6.3 Central nervous system tumours Grade I (non-infiltrating pilocytic astrocytoma) Grade II (well-to-moderately differentiated astrocytoma) Grade III (anaplastic astrocytoma) Grade IV (glioblastoma multiforme) Pituitary adenoma Craniopharyngioma Extragonadal germ cell tumour Primary cerebral lymphoma Choroid plexus tumours Ependymoma Oligodendroglioma Medulloblastoma Schwannoma Extradural meningioma Intramedullary ependymoma Astrocytoma Gliomas account for 50% of brain tumours and are divided into grade I (non-infiltrating pilocytic astrocytoma), grade II (well-to-moderately differentiated astrocytoma), grade III (anaplastic astrocytoma) and grade IV (glioblastoma multiforme). The prognosis deteriorates with rising tumour grade. In addition to these astrocytomas, there are other glial tumours including ependymomas that arise from ependymal cells (the epithelial like cells that line the ventricles of the brain and spinal cord), and oligodendrogliomas that arise from oligodendrocytes (the cells that make myelin sheaths). In the peripheral nervous system, neurofibromata (derived from non-myelin forming Schwann cells) and schwannomas (derived from Schwann cells that make myelin) are the most frequent glial tumours. Medulloblastoma is a glial tumour of childhood usually arising in the cerebellum, which may be related to primitive neuroectodermal tumours elsewhere in the CNS. Non-glial brain tumours include pineal parenchymal tumours, extragonadal germ cell tumours, craniopharyngiomas, meningiomas and choroid plexus tumours. Meningioma is the commonest non-glial tumour and constitutes 21% of brain tumours. The majority of spinal axis tumours in adults are extradural, metastatic carcinoma, lymphoma or sarcoma. Primary spinal cord tumours include extradural meningiomas (26%), schwannomas (29%), intramedullary ependymomas (13%) and astrocytomas (13%). Seventy per cent of primary brain tumours in adults are supratentorial, situated above the tentorium cerebelli, the tent of dura mater that lies between the cerebellum and the inferior portion of the occipital lobes. In contrast, primary brain tumours in children are usually located below the tentorium (Table 6.4). Glial tumours may produce both generalized and focal effects, and these will reflect the site of the tumour and the speed of its growth. General symptoms from the mass effect, increased intracranial pressure, oedema, midline shift and herniation syndromes are all seen, including progressive altered mental state and personality, headaches, seizures and papilloedema. Focal symptoms depend upon the location of the tumour (Figure 6.2). Although seizures are a feature of up to half of all glial tumours, fewer than 10% of first fits are due to tumours and only 20% of supratentorial tumours present with fits. Table 6.4 Brain tumours by age and site Figure 6.2 Common presentation of brain tumour by site. These tumours, which are more common in women, present as slowly growing masses producing headaches, seizures, motor and sensory symptoms and cranial neuropathies, depending on their site (Table 6.5 and Figure 6.3). Meningiomas are some of the few tumours that produce characteristic changes on plain skull X-rays with bone erosion, calcification and hyperostosis (excess growth of bone) (Figure 6.4). The sites of spinal axis tumours are 50% thoracic, 30% lumbosacral and 20% cervical or foramen magnum. These tumours present with radicular symptoms due to nerve root infiltration, syringomyelic disturbance (dissociated sensory loss of pain and temperature sensation) due to central destruction by intramedullary tumours, or sensorimotor dysfunction (limb weakness and a sensory level) due to cord compression. Neuroradiology has developed into the most important investigation in patients with suspected brain tumours, following the introduction of computed tomography (CT) in the mid-1970s by Geoffrey Hounsfield and magnetic resonance imaging (MRI) in the 1980s. Newer techniques, such as positron emission tomography (PET), single photon emission computerized tomography (SPECT) and functional MRI have also found roles in the diagnosis and management of patients with brain tumours. MRI with gadolinium enhancement is the imaging technique of choice with advantages over CT particularly for posterior fossa tumours and non-enhancing low-grade gliomas (see figures in Chapter 1). PET with fluorodeoxyglucose-18, which accumulates in metabolically active tissues, may help to differentiate tumour recurrence from radiation necrosis (Figure 6.4). Stereotactic biopsy is required to confirm the diagnosis, although occasionally tumours are diagnosed on clinical evidence, because biopsy might be hazardous, for example, in brain stem gliomas. Table 6.5 Clinical features of meningiomas by site Figure 6.3 CT scan image of meningioma. Figure 6.4 Co-registered and separate MRI and 18-fluorodeoxyglucose PET scan images from a patient with a paraventricular high-grade glioma, demonstrating high glucose utilization by the tumour. Some gliomas are curable by surgery alone and some by surgery and radiotherapy; the remainder require surgery, radiotherapy and chemotherapy, and these tumours are rarely curable. Surgical removal should be as complete as possible within the constraints of preserving neurological function. Radiation can increase the cure rate or prolonged disease-free survival in high-grade gliomas and may also be useful symptomatic therapy in patients with low-grade glioma, who relapse after initial therapy with surgery alone (Figure 6.5). Newer radiotherapy delivery techniques that have been pioneered in the treatment of brain tumours include both Gamma Knife and CyberKnife treatments, which have caught the attention and interest of the gently dozing neurosurgeon in the multidisciplinary team. Gamma Knife radiotherapy is delivered by 201 cobalt-60 sources arranged in a ring in a helmet that is bolted to the patient’s skull. CyberKnife radiotherapy uses a linear accelerator to deliver radiotherapy via a robotic arm that is linked to an image guidance system. CyberKnife radiotherapy does not need a skull frame because the real-time image linking means that if the patient moves, the robotic arm also moves so that the radiotherapy dose is delivered to the correct site. Figure 6.5 CT scan from a patient showing an Omaya shunt (a closed cerebrospinal fluid (CSF) shunt joining the lateral ventricle with a reservoir below the scalp) in place that is leaking, resulting in air seen in the right anterior brain and fluid accumulating around the shunt site. Omaya shunts may be placed to relieve non-communicating hydrocephalus caused by obstruction to CSF flow within the ventricular system, or to administer intrathecal chemotherapy. Chemotherapy with lipid-soluble nitrosoureas or temozolomide may prolong disease-free survival in patients with oligodendrogliomas and high-grade gliomas, although its high toxicity may not always merit this approach. One recent advance is the use of biodegradable wafers implanted with BCNU chemotherapy that may be inserted at the time of surgery. Gene therapy has been adopted for trials in gliomas with viral vectors being administered either into the blood or directly into the tumour by surgeons. The genetically modified viral vectors may be non-replicating viruses that deliver a transgene that causes an anticancer effect, or replicating oncolytic viruses that directly lyse cancer cells by replicating. Gliomas are a good model to try these methods because they are pretty much the only cells dividing in the brain (apart from microglia and endothelial cells). Similarly, anti-angiogenic therapies chiefly targeting vascular endothelial growth factor (VEGF) are used in clinical trials in high-grade glioma. Another line of research in gliomas has focussed on the development of immunotherapy targeting HER2 expression and autologous dendritic cell therapies. Similarly, some gliomas have activation of the AKT intracellular signalling pathway and this can be targeted by a number of drugs in development. The name AKT originates from the inbred strain of mouse that developed spontaneous lymphomas from which the AKT protein kinase was identified. Therapy of meningiomas is surgical resection, which may be repeated at relapse. Radiotherapy reduces relapse rates and should be considered for high-grade meningiomas or incompletely resected tumours. Relapse rates are 7% at 5 years if completely resected and 35–60% if incompletely resected. Unlike with other brain tumours, surgical resection does not have a useful role in primary cerebral lymphomas. In immunocompetent patients, the combination of chemotherapy and radiotherapy produces median survivals of 40 months. In contrast, in the immunocompromised patients, especially those with HIV infection, the prognosis is far worse, with a median survival of under 3 months. Palliative radiotherapy or best supportive care is the appropriate treatment options here. Early complications of cranial radiotherapy which occur in the first 3–4 months are due to reversible damage to myelin-producing oligodendrocytes. This recovers spontaneously after 3–6 months. It causes somnolence or exacerbation of existing symptoms in the brain and Lhermitte’s sign (shooting numbness or paraesthesia down the back and limbs precipitated by neck flexion) in the cord. Late complications include radiation necrosis, causing irreversible neurological deficits due to damage to blood vessels. This may mimic disease recurrence; it is radiation dose related and occurs in up to 15% of patients, with the highest frequency in children who have also received chemotherapy. PET scanning may differentiate radionecrosis and relapse. Table 6.6 Five-year survival rates of adult patients with brain tumours The prognosis of glial tumours depends upon the histology, the grade and size of the tumour, on the age and performance status of the patient and on the duration of the symptoms (Table 6.6). The median survival for anaplastic astrocytoma is 2–5 years, and for glioblastoma multiforme is 10–14 months. Meningiomas, if completely resected, are usually cured and the median survival is over 10 years. Case Study: The consultant’s mother.
Central nervous system cancers
Epidemiology
Aetiology
Percentageof all cancer
Rank of registration
Lifetime risk of cancer
Change in ASR(2000-2010)
5-year overall survival
Female
Male
Female
Male
Female
Male
Female
Male
Female
Male
Brain cancer
3
3
8th
10th
1 in 77
1 in 77
0%
+10%
16%
15%
Condition
Inheritance and genetics
Cutaneous manifestations
Eye
Nervous system
Brain tumours
Von Recklinghausen’s neuro-fibromatosis (NF-1)
Autosomal dominant NF-1 gene (encodes neurofibromin that regulates GTPases in signal transduction)
Café au lait macules, axillary freckles
Lisch nodules (pigmented iris hamartomas)
Neurofibromata
Schwann cell tumours of spinal and cranial nerves, meningiomas, ependymomas, optic gliomas
Acoustic neurofibromatosis (NF-2)
Autosomal dominant NF-2 gene (encodes Merlin protein involved in cell adhesion)
Café au lait macules less common than in NF-1
Presenile cataracts
Bilateral acoustic neuromas Neurofibromata
Schwann cell tumours of spinal and cranial nerves, meningiomas, astrocytomas, ependymomas, optic gliomas
Tuberous sclerosis
Autosomal dominant TSC1 gene (encodes hamartin protein) and TSC2 gene (encodes tuberin protein)
Adenoma sebaceum, Shagreen patches, subungual fibromata, café au lait spots
Seizures, mental retardation
Giant cell astrocytoma of the foramen of Munro, gliomas, ependymomas
Von Hippel–Lindau
Autosomal dominant VHL gene (encodes VHL protein involved in ubiquitination)
Skin hamartomas
Retinal angiomas
Cerebellar haemangioblastomas, ependymomas, pheochromocytoma
Pathology
Gliomas
Non-glial brain tumours
Astrocytomas
Meningioma
Other glial tumours
Primary spinal cord tumours
Presentation
Glial tumours
Adult
Child
Supratentorial
Supratentorial
Metastases
Craniopharyngioma
Glioma
Pinealoma
Meningioma
Optic glioma
Pituitary tumour
Infratentorial
Infratentorial
Metastases
Medulloblastoma
Acoustic neuroma
Cerebellar astrocytoma
Cerebellar haemangioblastoma
Ependymoma of fourth ventricle
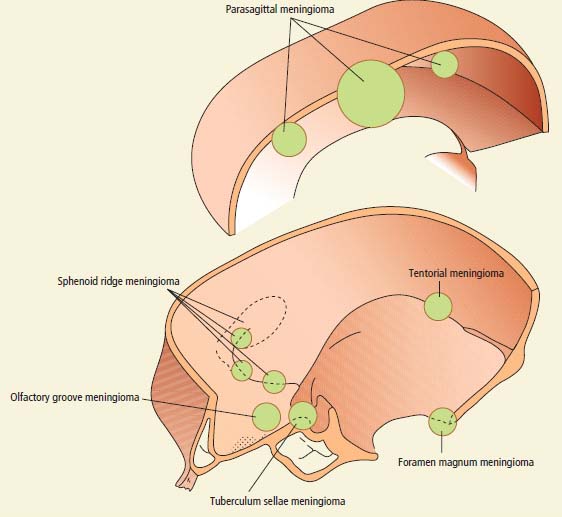
Meningioma
Spinal axis tumours
Investigation and staging
Tumour site
Clinical features
Parasagittal falx
Progressive spastic weakness
Numbness of legs
Olfactory groove
Anosmia
Visual loss
Papilloedema (Foster–Kennedy syndrome)
Frontal lobe syndrome
Sella turcica
Visual field loss
Sphenoid ridge
Cavernous sinus syndrome (medial)
Exophthalmos and visual loss (middle)
Temporal bone swelling and skull deformity (lateral)
Posterior fossa (foramen magnum, tentorium)
Hydrocephalus (tentorium)
Gait ataxia and cranial neuropathies
V, VII, VIII, IX and X (cerebellopontine angle)
Suboccipital pain, ipsilateral arm and leg weakness (foramen magnum)
Treatment
Complications of treatment
Tumour
5-year survival
Grade I glioma (cerebellar)
90–100%
Grade I glioma (other sites)
50–60%
Grade II (astrocytoma)
16–46%
Grade III (anaplastic astrocytoma)
10–30%
Grade IV (glioblastoma multiforme)
1–10%
Oligodendroglioma
50–80%
Meningioma
70–80%
Prognosis
 ONLINE RESOURCE
ONLINE RESOURCE
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


