Main causes
Inheritable defects
CPHDs
Pituitary transcription factor defects
LEPR or PROKR2 mutations
Isolated CH
TSHβ, TRHR or IGSF1 mutations
Invasive or compressive lesions
Craniopharyngiomas
Pituitary macroadenomas
Meningiomas or gliomas
Rathke cleft cysts
Empty sella
Iatrogenic causes
Cranial surgery or irradiation
Drugs (anti-cancer treatments, RXR agonists, dopamine)
Injuries
Head traumas
Traumatic delivery
Unusual causes
Vascular accidents
Pituitary infarction
Subarachnoid haemorrhage
Autoimmune diseases
Lymphocytic hypophysitis
Polyglandular autoimmune diseases
Infiltrative lesions
Iron overload
Sarcoidosis or Histiocytosis X
Infective diseases
Tuberculosis or Mycoses
9.2 CeH Diagnosis in Children
9.2.1 Inheritable CeH Forms
The various genes so far linked to CeH are illustrated in Fig. 9.1.
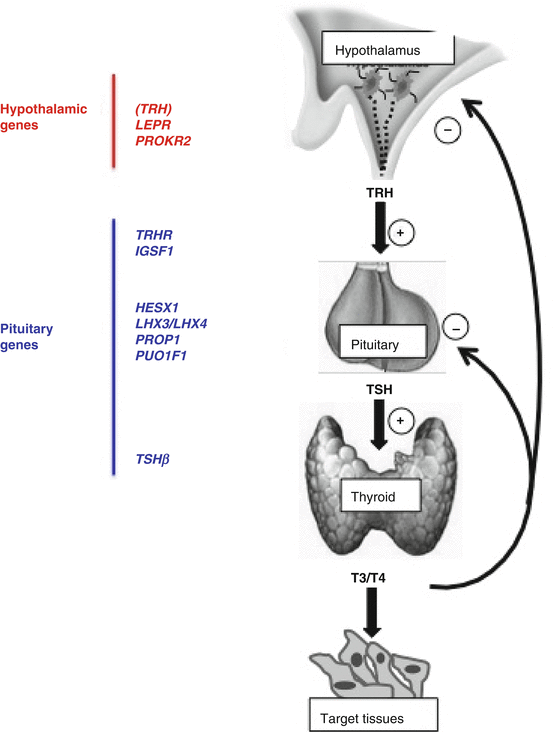
Fig. 9.1
Schematic illustration of the hypothalamic and pituitary genes so far associated with the pathogenesis of CeH. Though the involvement of TRH gene seems obvious, no variants have been so far detected in humans with CeH
In the neonates, CeH can be identified only by screening programs based on concomitant TSH and total T4 measurements in the dry blood spot [4–8]. CeH confirmation by serum FT4 and abnormal TSH response to TRH testing may reveal the risk of CPHDs and impending adrenal crisis [8]. CPHDs represent the more frequent forms of neonatal CeH. They can be the consequence of mutations in genes encoding for various pituitary transcription factors, such as PROP1, POU1F1 (or PIT1), HESX1, LHX3, or LHX4, or for hormone receptors relevant for several hypothalamic functions, such as LEPR or PROKR2 [1, 2, 9, 10]. In pituitary transcription factor defects, CeH can also have a delayed onset and be associated with hypoglycemia, craniofacial or pituitary abnormalities, and severity of growth retardation [1, 2, 9, 10]. The complex phenotypes associated with these defects are summarized in Table 9.2.
Table 9.2
Phenotypes associated with the inheritable forms of CeH
Genes (OMIM *gene number) | Associated phenotype and transmission (OMIM #disease number) |
|---|---|
TSHβ (*188540) | Recessively inherited severe isolated CeH of neonatal onset with high α-GSU, pituitary hyperplasia (#275100) |
TRH-R (*188545) | Recessively inherited isolated CeH with blunted TSH/PRL response to TRH and apparently uneventful infantile development, and with childhood (growth retardation) to adulthood onset |
IGSF1 (*300137) | X-linked CeH associated with low PRL, variable partial GH deficiency, and macrorchidism |
POU1F1 (*173110) | Moderate/severe CeH (dominant or recessive inheritance) of neonatal to infantile onset combined with GH and PRL defects, prominent forehead, mid face hypoplasia, depressed nose (#613038) |
PROP1 (*601538) | Recessively inherited moderate/severe CeH of neonatal to infantile onset, combined with GH, PRL, LH/FSH defects, and delayed ACTH deficiency, pituitary hypo-/hyperplasia (#262600) |
HESX1 (*601802) | Dominantly or recessively inherited panhypopituitarism associated with septo-optical dysplasia (SOD), supernumerary/hypoplastic digits (#182230) |
LHX3 (*600577) | Recessively inherited hypopituitarism with conserved ACTH function and associated with pituitary hypo- or hyperplasia, short/rigid cervical spine and variable deafness (#221750) |
LHX4 (*602146) | Dominantly inherited CPHD associated with abnormalities of cerebellum and small sella turcica (#262700) |
PROKR2 (*607123) | Variable CPHD associated with SOD or pituitary stalk interruption (SIP) (variable inheritance) |
LEPR (*601007) | Recessively inherited severe obesity and hyperphagia combined with delayed puberty and mild thyrotropin defect |
Inheritable forms of CeH due to biallelic TSHβ mutations are frequently associated with severe neonatal onset and characterized by the typical manifestations of congenital hypothyroidism (macroglossia, coarse cry, jaundice, failure to thrive and retarded growth, umbilical hernia, hypotonia, etc.). If untreated with l-thyroxine within 6 weeks of life, these patients generally develop cretinism [1, 6, 11, 12]. The association of CeH with high α-GSU levels in an infant is invariably indicative of a TSHβ defect [11].
The TRH knockout mice have a typical CeH phenotype [6], but no TRH gene defect has been documented so far in humans. However, a defective TRH action due to natural mutations in the TRHR gene has been so far described in two families [13, 14]. We reported a family with a complete TRH receptor defect caused by an early stop codon potentially leading to the translation of a truncated protein lacking all the seven transmembrane and intracellular domains [14]. The probands of this family represent a natural TRHR knockout model: a unique opportunity to understand the role of TRHR in humans. The early development of patients with complete TRH resistance appeared uneventful. The diagnosis in the male proband with homozygous TRHR mutations was reached because of delayed growth accompanied by fatigue at 11 years of age. The presence of this defect can be suggested by the blunted responses of TSH and PRL to TRH stimulation [13, 14] (Table 9.2). Unexpectedly, the same diagnosis was reached in the 33-year-old sister by genetic testing, during her second pregnancy. This woman with complete TRH resistance had reached her target height and normal IQ and has presently delivered three heterozygous babies with normal pre- and postnatal growth. In none of these cases, she experienced any lactating defect. Interestingly, when a hypothyroid questionnaire was administered to this woman before the start of l-thyroxine during the second gestation, she responded positively to only 1 question out of 12. Nevertheless, when the therapy was withdrawn for 6 weeks during her puerperium, the number of positive responses rose up to 10/12, thus indicating that thyroid hormone replacement had certain subjective beneficial effects that were unexpected a priori. Therefore, this study showed that the hypothalamic hormone is required to set the pituitary feedback mechanism at a level adequate to maintain free thyroxine levels in the normal range. In addition, the conservation of a significant nocturnal TSH surge in this condition indicates that TRH action influences the amplitude, but additional sleep-related factors account for the determination of the circadian oscillation. Interestingly, though TRH is also expressed in the pancreatic islets, we could not demonstrate any defect of glucose homeostasis in these patients [14].
Very recently, several familial cases of X–linked CeH from the Netherlands, the UK, and Italy have been reported to be associated with genetic defects in IGSF1 [15, 16]. This gene encodes a membrane protein containing immunoglobulin-like motifs but of still unclear biological functions that is expressed in the pituitary and testes. IGSF1 defects are associated with a novel syndrome including CeH and macroorchidism and seldom GH deficiency. CeH in these cases is associated with blunted TSH and PRL responses to TRH testing consistent with the finding of a reduced Trh-r expression in the pituitaries of IGSF1 knockout mice. Accordingly, IGSF1 transcripts were found in Pit1-dependent lineages (thyrotrope, lactotrope, and somatotrope). Igsf1-deficient male mice show low pituitary and serum TSH concentrations, decreased thyroxine and triiodothyronine concentrations, and increased body mass [15].
9.2.2 Acquired CeH Forms
The hypothyroid state is mild to moderate in most patients with acquired CeH, as the pituitary TSH reserve is infrequently depleted. Although manifestations of CeH are similar to those of primary hypothyroidism, they can be masked by symptoms of CPHDs [1, 2]. CeH represents a major false-negative result of the “reflex TSH strategy” for the diagnosis of thyroid dysfunction [1, 5–7]. Therefore, acquired CeH should be suspected in all subjects with known hypothalamic/pituitary lesions (e.g., craniopharyngiomas, pituitary macroadenomas) or in those with clinical and biochemical manifestations (e.g., growth retardation, fatigue, cholesterol elevation) suggestive of hypothyroidism despite normal/low circulating TSH. On serum samples, the diagnosis of CeH is usually suggested by the finding of low FT4 concentrations, associated with low/normal TSH levels [1, 2, 17, 18]. Nevertheless, some CeH patients with a predominant hypothalamic defect have high serum immunoreactive TSH levels but devoid of full biological activity. In these cases, TSH elevations are similar to those generally found in subclinical or mild primary hypothyroidism and may lead to the misdiagnosis [1, 2]. In Table 9.3, the conditions associated with low/normal TSH and low FT4 levels and that could come into differential diagnosis with CeH are listed.
Table 9.3
Conditions that can be associated with diminished FT4 serum levels and aberrantly low/normal TSH
Central hypothyroidism (hypothalamic hypothyroidism may seldom be associated with TSH values above the upper normal limit) |
Severe non-thyroidal illness (e.g., anorexia) |
L-T4 withdrawal syndrome |
Recovery from thyrotoxicosis |
Drugs inhibiting TSH secretion |
Allan-Herndon-Dudley syndrome (MCT8 mutations) |
TRα1 mutations |
When a low FT4 is combined with a normal TSH value, the diagnostic workup for the confirmation of CeH should include the exclusion of interference in FT4 or TSH measurements [1, 2]. In general, automated FT4 assays are less reliable than the equilibrium dialysis, which is however not compatible with the routine work. If interference is suspected, this should be explored by using a “two-step” assay or by mass spectrometry. If the problem persists, hormone measurement following equilibrium dialysis remains the gold standard for eliminating FT4 assay interference. Less frequently, TSH immunometric measurement can be interfered by the presence of heterophile antibodies in a patient’s serum, if directed against the same species as the assay antibodies: thus, a heterophile antibody that blocks TSH binding to either capture or detection antibodies will cause a falsely low TSH readout potentially indicating a central instead of a primary hypothyroidism. Though most of the manufacturers are nowadays providing reagents including the preimmune serum from the source animal, heterophile antibodies may still interfere in the TSH determination on some instances. If interference is suspected, the discordant TSH concentration should be checked (a) by means of an immunoassay using a different antibody pair, (b) after immunosubtraction by treatment with polyethylene glycol (PEG) or protein G, or (c) by dilution or recovery tests [1, 2].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


