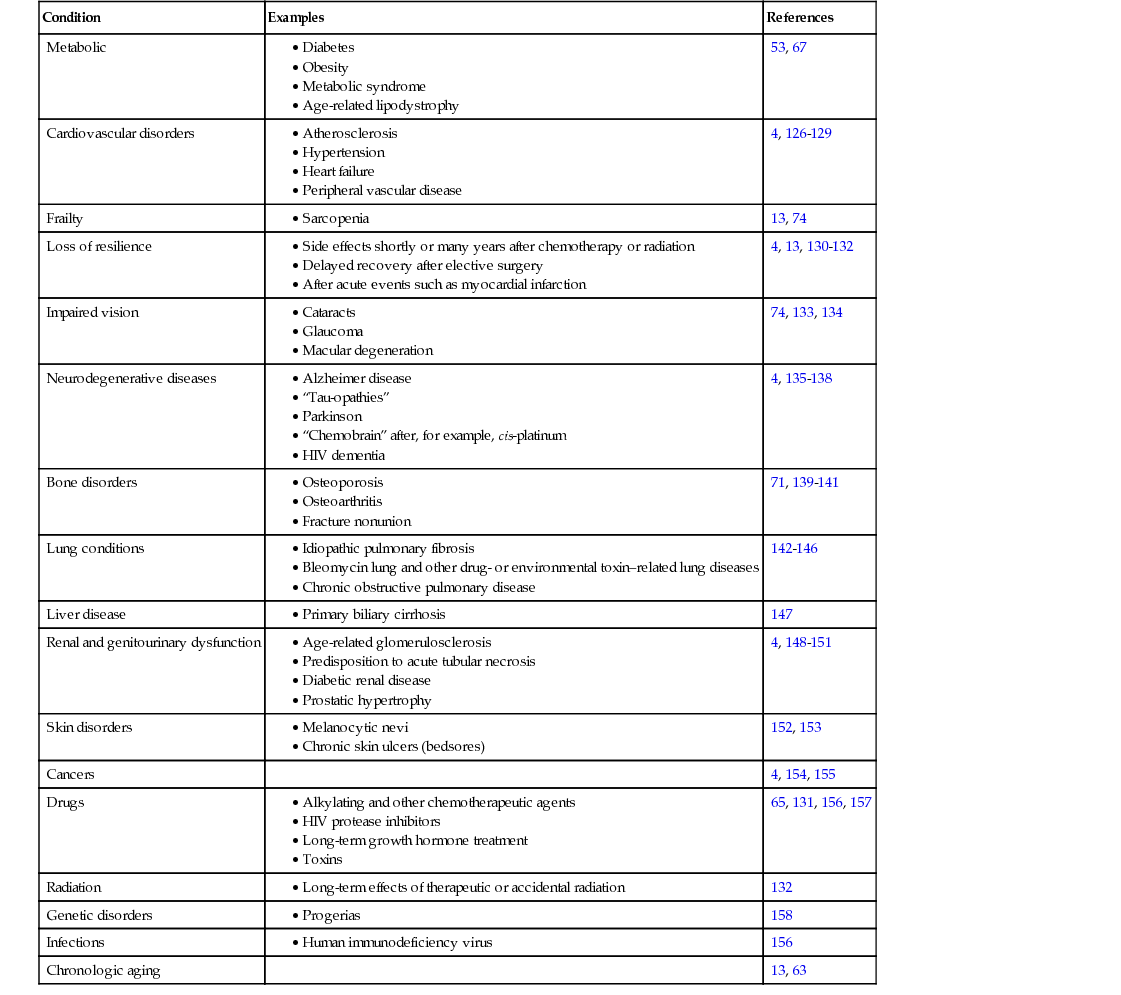
James L. Kirkland Aging changes are universal within a species and are intrinsic and progressive. They are universal; each true aging change should develop in all individuals in a species if they live long enough. Aging is intrinsic because these changes occur despite environmental cues, although the environment can alter their timing. The term progressive refers to the time dependency of aging processes. After adulthood, aging is associated with a general decline in cellular, tissue, and systemic function, loss of reproductive capacity, decreased resilience, and the ability to adapt to environmental perturbations and respond effectively to disease. Age-related phenotypes or diseases (e.g., development of certain cancers, such as prostate cancer, or onset of atherosclerosis) are likely predisposed to by fundamental aging processes that are operative during particular stages of life. These age-related phenotypes and diseases are heterogeneous and, unlike true aging changes, are not universal, occurring segmentally in different tissues and appearing at different times among individuals. Recent important advances have been made in our understanding of the basic biology of aging. These insights into the cellular and molecular biology of aging have led to development of a number of interventions, both lifestyle and drugs, that extend life span and health span—the period during life free of chronic disease, pain, disability, and dependency—at least in mice. If these can be translated into humans, such interventions may prove to delay, prevent, alleviate, or even reverse the age-related diseases and disabilities that are the leading drivers of morbidity, mortality, and health expenditures in developed and developing societies. These age-related conditions include atherosclerosis, most cancers, mild cognitive impairment, dementias, Parkinson and other neurodegenerative diseases, type 2 diabetes, renal dysfunction, arthritis, blindness, frailty, and sarcopenia. For each condition, chronologic aging is a major risk factor. Indeed, for most, aging is a larger risk factor than all others combined.1–4 Importantly, the major age-related diseases share the disturbances in tissue, cellular, and molecular function that also occur with chronologic aging. These include chronic sterile inflammation, cellular senescence, macromolecular damage (DNA, proteins, carbohydrates, and lipids), and stem and progenitor cell dysfunction. Based on these points, the geroscience hypothesis has been proposed: by targeting fundamental aging processes, it may be feasible to treat the major age-related chronic diseases and geriatric syndromes as a group, instead of one at a time. This hypothesis is being actively tested in experimental animal models and human cells. If true, and if the interventions that appear to be effective in targeting fundamental aging mechanisms in mice can be translated into humans, geriatrics practice and all of medicine as we know it could be transformed. The basic biology of aging field has moved from an era of descriptive research to hypothesis-driven research, with a focus on elucidating mechanisms and, most recently, into developing interventions that target fundamental aging processes. The next phase, which has already started, is to translate these interventions into clinical application. Modulators and interventions that delay age-related changes in experimental animals include caloric restriction, several hundred single-gene mutations across species and, most recently several drugs. The single-gene mutations that extend life span or health span involve the growth hormone (GH)–insulin-like growth factor-1 (IGF-1)–insulin signaling pathway and other pathways related to anabolism and caloric restriction, as well as inflammation, the renin-angiotensin system, and cellular senescence, among others. In general, these genetic and pharmacologic interventions are related to inflammation, cell survival, cellular senescence, macromolecular processing, fuel and metabolic sensing and processing, caloric restriction, and stem and progenitor cell function. These interlinked processes, which appear to be the most likely targets for future clinical interventions to delay age-related dysfunction and chronic diseases as a group, are considered in this chapter. A general decline in immunity occurs with aging, with increased susceptibility to infections, cancers, autoimmune disorders, and associated mortality. Immune cells undergo declines in function in aging organisms—for example, there is decreased macrophage function and impaired activation potential of T cells in older individuals.5–8 Additionally, the function of antiinflammatory pathways may also decline with aging, predisposing to development of a chronic, low-grade sterile inflammatory state that leads to tissue damage. The imbalance between proinflammatory and antiinflammatory pathways with aging has been termed inflammaging.9 Chronic, low-level, nonmicrobial (or sterile) inflammation develops in multiple tissues with both aging and age-related chronic diseases.9–12 The source of this age-related chronic inflammation has not been pinpointed precisely. Candidate mechanisms include dysregulation of the immune system, chronic antigenic stimulation (e.g., by latent viruses), oxidative stress, increases in dysfunctional macromolecules (e.g., unfolded or aggregated proteins, glycation end products, or reactive lipids), and accumulation of senescent cells (see later).9,13,14 Chronic inflammation can drive tissue dysfunction by at least two mechanisms. First, infiltrating immune cells can degrade tissues because they release reactive or toxic moieties. Second, inflammatory cytokines can provoke phenotypic changes in nearby cells that are independent of the immune system. For example, interleukin (IL)-6 and IL-8 can stimulate angiogenesis, disrupt cell-cell communication, impede macrophage function, induce innate immune responses, and promote epithelial and endothelial cell migration and invasion.6,15–21 Furthermore, increases in tissue inflammation under basal conditions may contribute to an increase in susceptibility to autoimmune diseases, as well as to a restricted capacity to boost the extent of inflammation further when needed. This restriction in the dynamic range of inflammatory and cellular stress responses likely constrains the capacity to respond appropriately to infection, immunization, or injury. Increases in inflammatory mediators, including elevated IL-6, tumor necrosis factor-α (TNF-α), and immune cell chemokine levels, are associated with multiple age-related diseases, including dementias,22 depression,23 atherosclerosis,24–28 cancers,29–31 and diabetes,32–34 as well as mortality.22,35,36 Sterile inflammation is perhaps the most important physiologic correlate of the age-related frailty syndrome37–40 that encompasses heightened vulnerability to stresses (e.g., surgery, infection, trauma), muscle wasting (sarcopenia), cachexia, and adipose tissue loss.37–39,41–49 Frailty predisposes to chronic disease, loss of independence, and mortality, as well as increased health costs.45,47 Cellular senescence refers to the essentially irreversible cell cycle arrest caused by potentially oncogenic and metabolic insults that evolved as a defense against tumor formation.13 Senescent cells adopt a characteristic enlarged shape, increased protein content, elevated tumor suppressor proteins such as p21CIP1 and p16INH4A, an increase in senescence-associated β-galactosidase activity, and elevated secretion of a number of growth factors, cytokines, immune cell–attracting chemokines, and matrix remodeling factors, collectively termed the senescence-associated secretory phenotype (SASP) or senescence-messaging secretome.50–52 A number of inducers, including oncogene activation, DNA damage, telomere erosion, oncogenic proteins, fatty acids, oxidative stress, mitogens, cytokines, and metabolites,52–55 can act alone or in combination to push cells into the senescent cell fate through pathways involving p16INK4A/Rb (retinoblastoma), p53/p21CIP1, and probably others.13 These contribute to the widespread changes in gene expression and chromatin remodeling (heterochromatin formation) that underlie senescence-associated growth arrest and changes in morphology. In these respects, cellular senescence can be viewed as a cell fate reminiscent of differentiation, replication, or apoptosis, with external and internal cues leading to activation of transcription factor cascades, gene expression changes, chromatin remodeling, and changes in function. Intracellular autocrine loops, including loops involving interleukins and reactive oxygen species (ROS), reinforce the progression to altered gene expression, irreversible replicative arrest, and heterochromatin formation over a matter of days to weeks. Cellular senescence contributes to age-related dysfunction and frailty and is frequently operative at sites of pathology underlying chronic age-related diseases.4,13,56 Senescence can occur at any point during life, even in blastocysts57 and the placenta.58 Indeed, senescence is important in remodeling during embryogenesis.59,60 Even though senescent cells are resistant to cell death through apoptosis,61 they are normally removed by the immune system in younger individuals.62 However, senescent cells accumulate in multiple tissues with advancing age.13,63,64 Senescent cell burden is, in turn, associated with life span. At 18 months of age, long-lived Ames dwarf, Snell dwarf, and GH receptor knockout mice have fewer senescent cells in their adipose tissue than age-matched control wild-type animals, whereas short-lived GH overexpressing mice have more.65 Caloric restriction sufficient to increase life span in mice is associated with decreased expression of p16INK4A, a senescence marker, in multiple tissues compared to animals fed ad lib.66 Conversely, senescent cells accumulate in fat and other tissues in obese animals and humans, especially when accompanied by diabetes.53,67 Consistent with the geroscience hypothesis, obesity and diabetes are associated with an accelerated onset of other aging- and senescence-associated conditions, including atherosclerosis, vascular dysfunction, sarcopenia, cognitive impairment, dementia, early menopause, and cancers, including non–hormone-dependent cancers.53,68,69 Progeroid mice, such as mouse models of Werner and Hutchinson-Guilford progerias, as well as Klotho-deficient, Ercc−/−, and BubR1H/H mice, have increased numbers of senescent cells.13,70–72 In comparisons across longer versus shorter lived mouse cohorts, senescent cell accumulation in liver and intestinal crypts predicted mean and maximum life spans.73 The SASP involves the release of proinflammatory cytokines, chemokines, prothrombotic factors, and extracellular matrix proteases that cause tissue damage, as well as extracellular matrix proteins that can contribute to dysfunctional tissue architecture. In addition to removing cells from the progenitor–stem cell pool, senescence may contribute to tissue dysfunction and chronic disease predisposition through the SASP and the chronic sterile inflammation and extracellular matrix disorganization that it causes. The associations among cellular senescence, aging, and age-related pathologies prompted testing if senescent cell clearance ameliorated dysfunction. Genetically targeting senescent cells in progeroid mice that expressed a drug-activatable so-called suicide gene only in senescent cells enhanced health span.74 Even clearing only around 30% of senescent cells from these mice led to partial reversal of age-related lipodystrophy and decreased progression of frailty, sarcopenia, and cataract formation.13,74 Furthermore, senescent cell removal later in life delayed progression of age-related pathology, even after it had emerged. Drugs that selectively eliminate senescent cells—senolytic drugs— have been discovered.75 These drugs alleviate age-related cardiac and carotid vascular dysfunction in old mice, radiation-induced muscle dysfunction in younger mice, and neurologic dysfunction, osteoporosis, and frailty in progeroid mice. Cellular senescence is associated with many of the chronic diseases and disabilities that are leading drivers of morbidity, mortality, and health costs.4,13,56 Senescent cells have been identified at sites of pathology in a number of these conditions and may have systemic effects that predispose to others (Table 9-1). These findings support a link between senescent cells and age-related dysfunction. There is now the prospect that drugs that target senescent cells and the SASP might come into clinical use to delay, prevent, ameliorate, or even reverse age- and senescence-related dysfunction and diseases in humans. TABLE 9-1 Conditions Associated with Cellular Senescence Aging is associated with the accumulation of damaged macromolecules, including DNA, proteins, carbohydrates, and lipids in and around cells. In most cases, these damaged macromolecules or the processes underlying their accumulation are related to chronic inflammation, cellular senescence, and stem and progenitor cell dysfunction, as well as to the major age-related chronic diseases. As considered later, many of the drugs that show promise for enhancing health or life span act on processes responsible for the generation or effects of these damaged macromolecules. Genomic damage accumulates over time due to environmental exposure and effects of metabolic byproducts, requiring DNA repair mechanisms to deal with these insults. Consistent with this, DNA repair and longevity are positively correlated.76 Premature aging phenotypes develop in mice with genetic ablation of genome repair mechanisms. Mitochondrial DNA damage, thought to be caused by ROS, may contribute to mitochondrial dysfunction with age, leading to inefficient adenosine triphosphate (ATP) production. Age-related mitochondrial dysfunction could play an important part in metabolic, cardiovascular, skeletal, and other age-related disorders.77 Genetic regulation at other levels can also become dysfunctional with aging—for example, at the level of noncoding RNA. Dysregulation of microRNAs (miRNAs), which have widespread effects on gene expression and cell function, and potentially also of proinflammatory noncoding RNAs, appears to be related to an age-related decline in Dicer protein, which processes miRNAs and other types of RNAs.78 Telomeres are structures at the ends of chromosomes, They comprise repeated TTAGGG nucleotide sequences in chromosomal DNA and several proteins that bind to telomeric DNA and cap the ends of chromosomes. During DNA replication, as cells divide, stretches of telomeric DNA at the ends of chromosomes, often 50 to 100 base pairs (bp) long, are lost at each division. Some cells, including germline, stem, cancer, and certain immune system cells, express telomerase, an enzyme complex that can regenerate telomeric DNA during each cell division, preventing telomere erosion. Loss of telomeric DNA alters the binding of proteins to telomeres and can result in unfolding or uncapping of chromosomal ends, which initiates cellular damage responses that can lead to loss of cellular replicative potential, loss of capacity to differentiate into specialized cell types, cell death, senescence, mitochondrial dysfunction with ROS generation, and inflammation. As individuals age, and cells have undergone increasing numbers of cell divisions, telomere erosion progresses and appears to contribute to tissue dysfunction and aging phenotypes. Accumulation of damaged, misfolded, abnormally glycated, oxidized, cross-linked, and aggregated proteins occurs in many tissues with aging and at sites of pathology in many age-related chronic diseases. Aggregated proteins contribute to the pathogenesis of many diseases, such as dilated cardiomyopathy, Alzheimer disease, Parkinson disease, insulin-dependent diabetes, and glomerulosclerosis.79 Slowed protein turnover and decreased protein clearance, which is normally promoted by proteasomal degradation and autophagy, occur with aging. Damaged proteins can induce cellular stress responses that can lead to inhibition of the capacity for progenitors to differentiate into specialized cells, cell death, inflammation, and cellular senescence. Degradation of damaged intracellular components by lysosomes or autophagy is part of the response to stress in many cells.80,81 By removing defective or damaged cellular components, autophagy contributes to cellular quality control. It is involved in cellular responses to proteotoxicity, genotoxicity, metabolic stress, and immune stress. Autophagy helps return cells to homeostasis by removing damaged or malfunctioning cellular structures, eliminating exogenous cellular damaging factors, or providing alternative sources of energy to help in resolving dysfunction by facilitating the turnover and recycling of essential constituents.82–84 It contributes to cell remodeling and differentiation by mediating the relative abundance of different proteins and eliminating damaged or excess cellular structures.85 It also contributes to immune responses and mitigates cellular damage.86,87 The central role for autophagy in regulating cellular function is disrupted in certain neurodegenerative disorders, leading to poor quality control of cellular components, in diabetes and obesity, resulting in altered energetic balance, and in autoimmune disorders and infections due to interference with immune responses.80,81,88 Blocking autophagy in muscle causes an accumulation of oxidized proteins, dysfunctional mitochondria, denervation, and decreased fiber force.89 Three autophagy pathways have been described in mammals—macroautophagy, chaperone-mediated autophagy, and microautophagy. During macroautophagy, targeted cytoplasmic cellular structures or macromolecules are isolated from the rest of the cell within a double-membraned vesicle, an autophagosome. The autophagosome then fuses with a lysosome, where its cargo is degraded and recycled. Chaperone-mediated autophagy is a process in which chaperone proteins selectively bind to dysfunctional cellular constituents and facilitate their importation and destruction by lysosomes. Macroautophagy and chaperone-mediated autophagy gradually become dysfunctional with aging.79,90,91 Effective autophagy has been associated with longevity and health span in genetic studies.79,90 Rapamycin extends life span and health span in mice.92 One mechanism may be through enhancing autophagy. Rapamycin inhibits the mammalian target of rapamycin (mTOR) protein, a kinase that promotes protein synthesis and inhibits autophagy. The activity of mTOR is increased in response to elevated amino acid availability, IGF-1, insulin, and other growth signals, whereas mTOR inhibition recapitulates some but not all of the effects of caloric restriction. A net effect of mTOR inhibition by rapamycin is an increase in protein quality, as well as decreases in the SASP and other effects. Clinical trials of rapamycin and related compounds are currently under way for a variety of age-related conditions and diseases, including dementias and cardiovascular diseases, and to enhance influenza vaccine responses in older populations.
Cellular Mechanisms of Aging
Introduction
Inflammation
Cellular Senescence
Condition
Examples
References
Metabolic
53, 67
Cardiovascular disorders
4, 126–129
Frailty
13, 74
Loss of resilience
4, 13, 130–132
Impaired vision
74, 133, 134
Neurodegenerative diseases
4, 135–138
Bone disorders
71, 139–141
Lung conditions
142–146
Liver disease
147
Renal and genitourinary dysfunction
4, 148–151
Skin disorders
152, 153
Cancers
4, 154, 155
Drugs
65, 131, 156, 157
Radiation
132
Genetic disorders
158
Infections
156
Chronologic aging
13, 63
Macromolecular Dysfunction
DNA
Telomeres
Accumulation of Dysfunctional Proteins
Autophagy
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cellular Mechanisms of Aging
9






