Langerhans cell histiocytosis (LCH) is heterogeneous disease characterized by common histology of inflammatory lesions containing Langerin + (CD207) histiocytes. Emerging data support a model in which MAPK activation in self-renewing hematopoietic progenitors may drive disseminated high-risk disease, whereas MAPK activation in more differentiated committed myeloid populations may induce low-risk LCH. The heterogeneous clinical manifestations with shared histology may represent the final common pathway of an acquired defect of differentiation, initiated at more than one point. Implications of this model include re-definition of LCH as a myeloid neoplasia and re-focusing therapeutic strategies on the cells and lineages of origin.
Key points
- •
Dendritic cells (DCs) are immune cells that arise from different lineages with a shared function of presenting antigen and activating adaptive immunity.
- •
Langerhans cell histiocytosis (LCH) arises from myeloid DC precursors. Mitogen-activated protein kinase (MAPK) activation is a universal feature of CD1a + langerin + LCH cells. The clinical extent of LCH is related to the stage of development in which somatic MAPK mutations arise, either self-renewing progenitors or committed precursors.
- •
Activating MAPK mutations in hematopoietic stem cells and committed myeloid precursors support classification of LCH as a myeloid neoplasia.
Introduction to Langerhans cell histiocytosis
The Histiocytoses
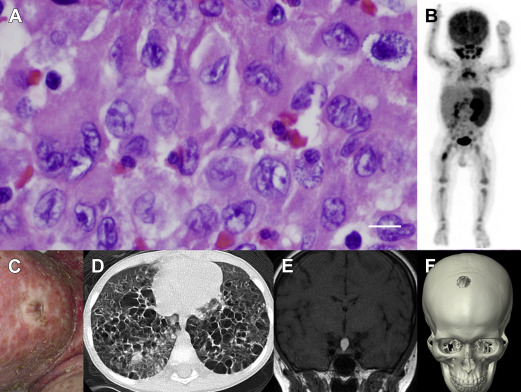
The spectrum of histiocytic diseases is characterized by collections of abnormal histiocytes or, literally, tissue cells related to myeloid cells of the mononuclear phagocyte system (MPS). LCH is defined by the presence of a large pale-staining histiocyte with high expression of CD1a and langerin (CD207) and containing Birbeck granules ( Fig. 1 ). These features, shared with Langerhans cells (LCs) of the epidermis, are the basis of classifying the disease as an LCH. Prior to this, LCH was known as histiocytosis X, a disease entity incorporating historically described syndromes: Hand-Schüller-Christian disease, characterized by lytic bone lesions and mucosal lesions; Letterer-Siwe disease, a fatal hepatosplenomegaly; and eosinophilic granuloma of bone. The discovery of the Birbeck granule in LCs in 1961 and identification of the same organelle in histiocytosis X by Nezelof and colleagues in 1965 formed the basis for modern models of LCH. Another influence driving this model of LCH at the same time was the model of the MPS in which peripheral macrophages were thought to be continually renewed from bone marrow–derived monocytes. LCs had recently joined the MPS by virtue of their expression of MHC class II, complement and Fc receptors, and repopulation by bone marrow–derived cells after transplantation. The formation of LCH was, therefore, perceived to be an aberration of this development, leading to the accumulation of abnormal LC-like cells in inappropriate locations.
Clinical Overview
The clinical spectrum of LCH ranges from a trivial single lesion to aggressive and potentially lethal disseminated disease (see Fig. 1 ). Almost every organ system has been reported to be involved by LCH lesions. In infants, skin lesions are common and may represent skin-limited disease that resolves without therapy; alternatively, skin rash may be part of a constellation of lesions in disseminated multisystem disease. In older children and adults, presenting symptoms may include pain from bone lesions, dyspnea from lung lesions, failure to thrive from intestinal involvement, or uncontrolled thirst from pituitary infiltration and resulting diabetes insipidus. The range of presenting symptoms and overlapping clinical features with more common childhood conditions make LCH a challenging diagnosis. Once biopsy is performed, however, the unique features of LCH cells and the inflammatory context of lymphocytes, eosinophils, and macrophages are pathognomonic. Although the number of pathologic histiocytes and relative contribution of infiltrating leukocyte populations may be variable, the overall pattern is conserved across the spectrum of clinical manifestations of LCH (see Fig. 1 ).
Rationale for Current Approaches to Langerhans Cell Histiocytosis
Clinically, LCH has been categorized as having a high risk of mortality when it involves bone marrow, spleen, or liver or as low risk in any other site. Treatment regimens are empiric, as summarized by Arceci and colleagues in the last Hematology/Oncology Clinics of North America issue dedicated to LCH (1998): “The variety of different treatment approaches to such patients has prompted some individuals to believe that LCH treatment ‘strategy’ is based more on a roulette wheel than on scientifically based logic. Certainly, part of the confusion and lack of consensus is derived from a persisting ambivalence as to whether LCH is primarily a neoplastic disorder, an immunodysrgulatory disorder, or a disorder with characteristics of both.” Vinblastine and prednisone have been the standard induction therapy for decades, although LCH-II and LCH-III trials demonstrated improved outcomes with dose intensification and therapy prolongation.
Introduction to Langerhans cell histiocytosis
The Histiocytoses
The spectrum of histiocytic diseases is characterized by collections of abnormal histiocytes or, literally, tissue cells related to myeloid cells of the mononuclear phagocyte system (MPS). LCH is defined by the presence of a large pale-staining histiocyte with high expression of CD1a and langerin (CD207) and containing Birbeck granules ( Fig. 1 ). These features, shared with Langerhans cells (LCs) of the epidermis, are the basis of classifying the disease as an LCH. Prior to this, LCH was known as histiocytosis X, a disease entity incorporating historically described syndromes: Hand-Schüller-Christian disease, characterized by lytic bone lesions and mucosal lesions; Letterer-Siwe disease, a fatal hepatosplenomegaly; and eosinophilic granuloma of bone. The discovery of the Birbeck granule in LCs in 1961 and identification of the same organelle in histiocytosis X by Nezelof and colleagues in 1965 formed the basis for modern models of LCH. Another influence driving this model of LCH at the same time was the model of the MPS in which peripheral macrophages were thought to be continually renewed from bone marrow–derived monocytes. LCs had recently joined the MPS by virtue of their expression of MHC class II, complement and Fc receptors, and repopulation by bone marrow–derived cells after transplantation. The formation of LCH was, therefore, perceived to be an aberration of this development, leading to the accumulation of abnormal LC-like cells in inappropriate locations.
Clinical Overview
The clinical spectrum of LCH ranges from a trivial single lesion to aggressive and potentially lethal disseminated disease (see Fig. 1 ). Almost every organ system has been reported to be involved by LCH lesions. In infants, skin lesions are common and may represent skin-limited disease that resolves without therapy; alternatively, skin rash may be part of a constellation of lesions in disseminated multisystem disease. In older children and adults, presenting symptoms may include pain from bone lesions, dyspnea from lung lesions, failure to thrive from intestinal involvement, or uncontrolled thirst from pituitary infiltration and resulting diabetes insipidus. The range of presenting symptoms and overlapping clinical features with more common childhood conditions make LCH a challenging diagnosis. Once biopsy is performed, however, the unique features of LCH cells and the inflammatory context of lymphocytes, eosinophils, and macrophages are pathognomonic. Although the number of pathologic histiocytes and relative contribution of infiltrating leukocyte populations may be variable, the overall pattern is conserved across the spectrum of clinical manifestations of LCH (see Fig. 1 ).
Rationale for Current Approaches to Langerhans Cell Histiocytosis
Clinically, LCH has been categorized as having a high risk of mortality when it involves bone marrow, spleen, or liver or as low risk in any other site. Treatment regimens are empiric, as summarized by Arceci and colleagues in the last Hematology/Oncology Clinics of North America issue dedicated to LCH (1998): “The variety of different treatment approaches to such patients has prompted some individuals to believe that LCH treatment ‘strategy’ is based more on a roulette wheel than on scientifically based logic. Certainly, part of the confusion and lack of consensus is derived from a persisting ambivalence as to whether LCH is primarily a neoplastic disorder, an immunodysrgulatory disorder, or a disorder with characteristics of both.” Vinblastine and prednisone have been the standard induction therapy for decades, although LCH-II and LCH-III trials demonstrated improved outcomes with dose intensification and therapy prolongation.
Molecular insights into pathogenesis of Langerhans cell histiocytosis
Langerhans Cell Histiocytosis: The Debate
The fundamental nature of LCH as neoplastic versus reactive disorder has been an ongoing debate. The granulomatous histology with quiescent histiocytes suggested potential autoimmune or infectious etiology but the unique appearance of LCH cells and destructive nature of lesions hinted at dysplastic development. Although Nezelof and Basset described LCs as the stem cell of LCH, they also acknowledged the prevailing view that elements of the MPS, including LCs, were continually replenished by the differentiation of bone marrow–derived precursors. Many hypotheses emerged that LCH might arise from LC precursors in a state of arrested development, misguided to inappropriate sites by a pathologic cytokine, or chemokine milieu, but no unifying extrinsic explanation for pathologic LCH cell differentiation was ever achieved (reviewed by Laman and colleagues ). A neoplastic origin for LCH was suggested by the coincidence of LCH with myelodysplastic syndrome and other malignancies and a major breakthrough came with the finding the LCH cells are clonal. Persistent failure to identify genetic abnormalities in systematic analysis of LCH lesions, however, tempered classification of LCH as a cancer.
Somatic Mitogen-Activated Protein Kinase Mutations in Langerhans Cell Histiocytosis
In 2010, Rollins and colleagues reported the seminal finding of recurrent BRAF V600E point mutations in approximately 60% of LCH lesions. BRAF is a central kinase that transduces signals through the MAPK pathway, which regulates numerous essential cellular functions ( Fig. 2 A). The BRAF mutation encoding the V600E substitution leads to constitutive activation of downstream mitogen-activated protein kinase kinase (MEK) and extracellular signal-related kinase (ERK) and is observed at high frequency in melanoma, in approximately 7% of human cancers overall, and in several benign neoplastic conditions, including epidermal nevi and colon polyps. Subsequently, whole-exome sequencing of LCH lesions has revealed recurrent mutations in MAP2K1 (encoding MEK1) in another 20% of patients and cases of mutations in other MAPK pathway genes, ARAF and ERBB3 , on a low overall mutation rate background (0.03 mutations per megabase) (see Fig. 2 ). These mutations all induce ERK activation, a universal feature of LCH cells. The genomic landscape of LCH is discussed in detail elsewhere in this issue.
Branches of dendritic cell differentiation
The insight of Nezelof and Basset identified commonality between LCs and LCH but by their own reckoning could not fully explain the histogenesis of LCH. The identification of MAPK pathway mutations provides a genetic neoplastic etiology and an important investigational tool with which to track potential LCH precursor cells. The problem of understanding exactly how potential LCH precursors differentiate abnormally remains, however, unsolved.
The Phenotype of Langerhans Cell Histiocytosis Dendritic Cells
To understand how aberrant DC differentiation might give rise to LCH cells, it is necessary to consider the phenotype of LCH cells in more detail. Breathnach and colleagues noted that in addition to Birbeck granules, acid phosphatase, nonspecific esterase, and α-D mannosidase were expressed by LCs and LCH cells. The advent of monoclonal OKT6, binding CD1a was a major advance both conceptually and diagnostically as was the description of langerin. Expression of CD1a is universal in LCH, and typically CD1a and langerin are highly coexpressed. Birbeck granules are associated with high CD1a and langerin and are most abundant in the skin but may be absent in the liver and in old xanthomatous lesions. Similarly, several reports describe heterogeneous states of differentiation of LCH cells within lesions, including variable CD1a + /langerin subpopulations. Antibody-recognizing BRAF V600E (VE1) has also demonstrated that the mutation is present in some langerin− cells within LCH lesions.
Despite these similarities, gene expression studies have demonstrated many differences between LCH cells and LCs: LCH cells express relatively less epithelial cell adhesion molecule (EpCAM), epithelial (E)-cadherin, and CD36; they also express higher levels of CD2, CD11b, CD11c, CD13, CD33, CD66c, and CD300LF, markers associated with myeloid DCs at all states of maturation. LCH cells also express high levels of cytokines and chemokines, including SPP1 (osteopontin), together with receptors CCR1 and NRP1 that influence capacity to recruit and interact with T cells. When isolated from lesions, they are somewhat immature, promoting a T h 2-rich environment enriched with eosinophils and regulatory T cells. These observations support a hypothesis that aberrant differentiation of a myeloid progenitor leads to a series of LC-like differentiation steps that are time and location dependent. The nature of this precursor remains undefined although the low expression of S100A8/9 indicates that LCH cells are unlikely to be recently derived from monocytes.
Origin and Homeostasis of Langerhans Cells in the Steady State
It is now appreciated that LCs are unique not only in their residence in the epidermis and mucosae but also in their origin and homeostasis. Contrary to the MPS model, LCs are not continually replenished from blood-borne progenitors in the steady state. The first LCs are derived from myeloid precursors during fetal life, which differentiate under the influence of the colony stimulating factor 1 receptor (CSF1R) cytokine interleukin (IL)-34 and autocrine production of transforming growth factor (TGF)-β. Ultimately, fetal LC progenitors are derived from primitive waves of hematopoiesis that are c-myb and flt3 independent and precede definitive hematopoiesis in the fetal liver. Once seeded in the epidermis, LCs are able to undergo homeostatic self-renewal, as first suggested in humans and proved by elegant experiments in mice. The independence of LCs from monocytes and blood DCs in the steady state is also demonstrated by the preservation of these cells in patients with genetic deficiency of monocytes and DCs and in patients who have received limb transplants. The development and subsequent self-renewing potential of a tissue macrophage/DC network during fetal life raises new possibilities for the origin of LCH cells. It is conceivable that somatic mutation could arise in these cycling cells and could result in LCH or that activation of a more widely distributed fetal precursor could occur (see Fig. 2 B).
Replenishment of Langerhans Cells During Inflammation
Although fetal-derived LCs are self-renewing in epidermis during steady state, bone marrow–derived precursors are recruited during inflammation, such as graft-vs-host disease after human stem cell transplantation. In mice, the epidermis is initially infiltrated by classical monocytes, but a second wave of more long-lived LC precursors has recently been described. In a recapitulation of embryonic development, IL-34 is also required to maintain long-term LC repopulation.
Dual Inflammatory Langerhans Cell Precursors
The duality of inflammatory LC precursors in mice also finds resonance with in vitro experiments in humans. A 2-phase kinetic was observed many years ago in serial skin biopsies of delayed-type hypersensitivity reactions. Langerin + cells can be derived from monocytes, from CD14 + cells appearing in CD34 + cultures, and from dermal CD14 + cells that are now known to be monocyte derived. All these may represent the monocyte pathway of short-term recruitment. In addition, CD1a + CD14 − intermediates with restricted LC potential can be generated from CD34 + progenitors. This suggests potential for an alternative pathway of LC differentiation, a conclusion that was recently supported by direct comparisons of CD14 monocytes and CD1c + blood DCs exposed to LC differentiation conditions. In these experiments, surprisingly, CD1c + DCs expressed much higher levels of CD1a and langerin than monocytes and only CD1c + DCs rapidly formed Birbeck granules. Either granulocyte-macrophage colony-stimulating factor or thymic stromal lymphopoietin was able to induce CD1a expression, and high langerin was promoted by TGF-β or bone morphogenetic protein 7. The role of IL-34 was not explored. Together these results suggest that the DC differentiation pathway may contribute to long-term LC precursors observed in mice and, furthermore, that both bone marrow–derived monocytes and myeloid DCs can express langerin and are candidate precursors for LCH cells.
Langerhans Cells Are Not the Only Fruit: Other Human Dendritic Cells
LCs are the paradigmatic migratory DCs, but blood and interstitial tissues contain 2 other populations of myeloid DCs: a minor subset of CD141 + cells and a major subset of CD1c + cells ( Fig. 3 ). Representatives of both are found in the blood and lymph nodes and are evolutionarily conserved in mammals, corresponding to the 2 subsets of classical or conventional DCs described in mice. The term, myeloid , is specific in humans and refers to the expression of antigens typically seen on granulocytes or monocytes, including CD13, CD33, CD11b, and CD11c. Plasmacytoid DCs typically lack myeloid antigens and are morphologically and functionally distinct, providing a major source of type 1 interferon in response to viral infection (reviewed by Reizis and colleagues ). They are typically found only in lymph nodes and inflamed tissues. In addition, several monocyte-derived DCs and macrophages are found in human tissues.
Related posts:
 Dedication
Dedication
 Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis
Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis
 Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
 Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


