Fig. 18.1
The pro-inflammatory/suppressive phenotype of TERS-imprinted myeloid cells. The characteristics of TERS-imprinted bone marrow-derived macrophages (middle) are compared with those of canonical characteristics of M1 (left) and M2 (right) macrophages (Adapted from Zanetti (2013))
Taking into account the current evidence, the cell-extrinsic effects of the tumor UPR appear to be contrasting: on the one hand promoting immune surveillance of hyperploid tumor cells via ER stress-enforced calreticulin expression (Senovilla et al. 2012), while, on the other, imprinting macrophages and dendritic cells with a pro-inflammatory/suppressive, mature phenotype with functional abnormalities with respect to antigen processing and presentation to T cells, as will be discussed below. To reconcile these seemingly contrasting effects, we suggested (Mahadevan et al. 2013) that the tumor UPR response may fulfill both functions, perhaps promoting cellular immunity against hyperploid cells on the one hand early during tumorigenesis, while ultimately undermining the immune response against cancer cells. That clinical tumors samples exhibit heterogeneous ploidy (Ohyama et al. 1990), and tetraploidy is key event in the progression of diverse histological subtypes (Davoli and de Lange 2011), suggest that this might indeed be the case. The seemingly Janus-faced cell-extrinsic role of the tumor UPR is shown in Fig. 18.2 (adapted from (Mahadevan et al. 2013)).
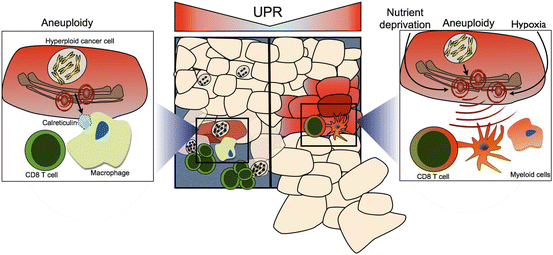
Fig. 18.2
Janus-faced effects of the tumor UPR on anti-tumor T cell immunity. (Left and left inset) Hyperploid cancer cells induce a UPR-dependent translocation of calreticulin to the cell surface where it serves as a “eat-me” signal for phagocytic cells (macrophages and dendritic cells). These take up tumor cell debris and apoptotic bodies, and present tumor antigens to T cells, inducing an anti-tumor immune response, which leads to the selective elimination of hyperploid cancer cells (Senovilla et al. 2012). (Right panel and right inset) The tumor UPR polarizes infiltrating myeloid cells to a pro-inflammatory/suppressive phenotype characterized by inefficient antigen presentation and CD8 T cell cross-priming, ultimately derailing anti-tumor T cell immunity, leading to tumor growth facilitation (Mahadevan et al. 2012) (From Mahadevan et al. ( 2013 ) with permission)
18.5 TERS Inhibits Antigen Presentation and CD8 T Cell Priming by Bone Marrow-Derived DC
In a series of experiments, we demonstrated that TERS impacts adversely upon myeloid DC cross-presentation and cross-priming (Mahadevan et al. 2012), two events associated with the induction of CD8 T cell-mediated immunity.
18.5.1 Effects on Cross-Presentation
To study cross-presentation we used a system in which the ovalbumin (OVA) SIINFEKL peptide bound to the H2-Kb molecule can be detected by flow cytometry using a monoclonal antibody. Reproducibly, OVA-fed, TERS-imprinted DC have reduced display of the SIINFEKL/H2-Kb complex at the cell surface, while the expression of MHC Class I molecules remains constant or even increased over that of OVA-fed control DC. Thus, TERS down-regulates cross-presentation.
18.5.2 Effects on Cross-Priming/T Cell Activation
To study cross-priming we used CD8 T cells from OT-I mice whose T cell receptor (TCR) is specific for the SIINFEKL/H2-Kb complex. In this model, OVA-fed bone marrow-derived DC efficiently induce both the activation and proliferation of OT-I CD8 T cells. When OT-I CD8 T cells are co-cultured with OVA-fed TERS-imprinted bone marrow-derived DC, however, while being activated, they proliferate poorly. On average, the majority (>70 %) of CD8 T cells are activated/non-dividing T cells. PD-1, a marker of “exhausted” T cells, is not upregulated. Importantly, antigen-specific CD8 T cells activated by TERS-imprinted myeloid DC show decreased ability to kill target cells [our unpublished data].
Importantly, we found that TERS-imprinted bone marrow-derived DC could exert dominant suppression over cross-priming by normal bystander antigen presenting cells. When TERS-imprinted bone marrow-derived DC, with or without antigen, are added to co-cultures naïve CD8 T cells and antigen-fed control bone marrow-derived DC, the proliferation of CD8 T cells is suppressed.
18.5.3 Mechanisms of Cross-Priming Defect
Efforts to restore the proliferative defect showed the following. (i) The addition of excess SIINFEKL antigen (1 μg/mL), rescued proliferation in OT-I T cells cross-primed by TERS-imprinted bone marrow-derived DC. (ii) The addition of exogenous IL-2 during cross-priming failed to rescue OT-I T cell proliferation, ruling out the possibility of classical anergy (Beverly et al. 1992). (iii) Removal from the co-culture containing TERS-imprinted bone marrow-derived DC partially restored T cell proliferation, although with fewer cell divisions, suggesting the importance of cell-cell contact. (iv) Whereas the addition of exogenous L-arginine to the co-culture did not improve T cell proliferation, the addition of L-norvaline, a competitive inhibitor of arginase, rescued it in great part (80 %). Taken together, these results suggest that tumor UPR-mediated myeloid cell-derived arginase activity and impaired cross-presentation together contribute to the T cell proliferative defect observed. Interestingly, however, addition of L-norvaline did not rescue T cell proliferation caused by dominant suppression.
18.5.4 TERS-Imprinted Myeloid DC in Context
Modeling the cell-extrinsic influence of the tumor UPR showed that TERS-imprinted BMDC are phenotypically mature, upregulate costimulatory molecules, have diminished cross-presentation capacity, and exert suppressive activity over CD8 T cells and bystander DC. Tolerogenic DC have been described in various systems (Steinman et al. 2003), and were initially defined in the periphery as steady-state, immature cells able to present antigen that suppress T cell activity because of inadequate co-stimulatory capacity (Gabrilovich et al. 1997; Steinman and Nussenzweig 2002). In the microenvironment of solid tumors of several histological types, infiltrating dendritic cells can be identified that display an immature phenotype with decreased MHC Class II, CD80, CD86, and CD83 expression, with presumed passive T cell inhibitory activity (Chaux et al. 1997; Bell et al. 1999; Pinzon-Charry et al. 2005; Tesone et al. 2013).
However, evidence has begun to accumulate ascribing active immunosuppressive activity via several mechanisms (e.g. arginase, IDO, and PD-L1 activity; for review, see (Tesone et al. 2013)) to phenotypically mature, so-called “regulatory” dendritic cells (Tesone et al. 2013). TERS-imprinted myeloid DC recapitulate ab initio several characteristics of these cells, including increased CD80, CD86, PD-L1, MHC Class II, and arginase activity with decreased antigen presentation capacity (Stoitzner et al. 2008; Liu et al. 2009; Norian et al. 2009; Scarlett et al. 2012; Engelhardt et al. 2012; Tesone et al. 2013). Given that regulatory dendritic cells have been isolated from epithelial cancers prone to a microenvironmental UPR, it is possible that the tumor UPR is a key modulator of myeloid antigen presenting cell, and ultimately, T cell function. A comparison of TERS-imprinted myeloid-derived dendritic cells with TIDC in different murine experimental systems and human patients is presented in Table 18.1.
Table 18.1
Comparison of different types of tumor associated myeloid cells and their effects on T cells
Type of cells | Mechanism of origin | Effect on CD8 T Cells | Mechanism of action | CTL Function | Ref. |
|---|---|---|---|---|---|
MDSC (Gr1+) | Unknown | Inhibit naïve cell expansion | Immature DC | ND | (Kusmartsev et al. 2004) |
TCR nitration | |||||
TuDC (CD11c+) | Unknown | Inhibit naïve cell expansion | Arginase and rescued by norvaline | ND | (Norian et al. 2009) |
Dominant suppression | |||||
TuDC (CD11c+) | Unknown | Inhibit naïve cell expansion and Ag experienced cell re-stimulation | Ag experienced CD8 T cells rescued by TLR7/TLR9 ligands | Decreased | (Engelhardt et al. 2012) |
Reg DC (MHC II/CD11c+/CD86+) | Tumor-derived PGE2, TGF-β1 | Inhibit tumor Ag experienced T cell re-stimulation | PD-L1, arginase? | ND | (Scarlett et al. 2012) |
Dominant suppression | |||||
BMDC* (CD11c+) | Transmissible UPR | Inhibit naïve cell expansion | Reduced Ag presentation | Decreased | (Mahadevan et al. 2012) |
Dominant suppression | Arginase and rescued by norvaline |
18.6 Implications of TERS-Directed Cross-Priming on Fate Determination of CD8 T Cells
Initial lineage analysis of CD8+ T cells cross-primed by TERS-imprinted bone marrow derived DC showed transcriptional upregulation of the cytokines Il–10 and Tnf- α but not Il–17, upregulation of Foxp3, and downregulation of the costimulatory molecule CD28. LAG3, a negative costimulatory molecule (Huard et al. 1994) found on tumor-infiltrating T cells (Grosso et al. 2007), was slightly up-regulated. When we analyzed the 96-h TERS-imprinted myeloid DC:T cell co-culture supernatant, we observed increased secretion of IL-2 but no elevation of IL-10, IL-17, IFN-γ or TNF-α compared to control (Fig. 18.3). A provisional conclusion is that CD8 T cells cross-primed by TERS-imprinted bone marrow-derived DC display an uncommitted phenotype with potential suppressive characteristics (CD28 downregulation and Il–10 upregulation) (Filaci et al. 2007). Surprisingly, CD8+ T cells cross-primed by TERS-imprinted BMDC also demonstrated disproportionately high splicing of Xbp–1 compared to only modest upregulation of other UPR elements, the significance of which remains unknown.
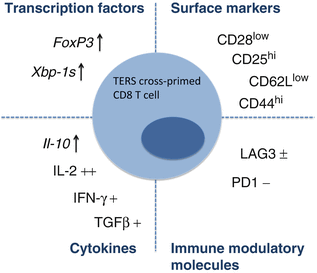
Fig. 18.3
Synopsis of phenotypic characteristics of murine CD8 T cells cross primed by TERS-imprinted myeloid DC
In sum, the phenotype of CD8+ T cells cross-primed by TERS-imprinted myeloid DC appears similar to that of CD8+/CD28– regulatory T cells secreting IL-10 and TNF-α, and expressing FOXP3, which have been found to infiltrate a variety of human tumors (Becker et al. 2000; Kruger et al. 2001; Filaci et al. 2007; Mahic et al. 2008). It still remains to be seen whether, like human CD8 suppressor T cells, TERS-directed CD8 T cells have suppressor functions effected by surface ecto-ATPases (e.g. CD39) and/or soluble mediators (e.g. IL-10). A comparison of the CD8+ T cell phenotype derived from TERS-imprinted APC with CD8+T cells infiltrating human tumors is presented in Table 18.2.
Table 18.2
Comparison of tumor-associated CD8 regulatory T cells
Immune phenotype | Species | Mechanism of Origin | Inhibitory Effects | Mechanism of Action | Ref. |
|---|---|---|---|---|---|
CD8+/CD28-/CD45RA+/CCR7-/FOXP3-/CD56- | Human | Soluble factors (IL-2, IL-10, TGF-b, GM-CSF) | Suppression of allogeneic CD8 and CD4 T cell expansion | CD39 | |
Agnon-specific in vitro | IL-10 | ||||
CD8+/CD45RO+/CCR7+/IL-10+ | Human | Tumor-associated plasmacytoid DC | Suppression of Ag-specific and allogeneic T cell activation | IL-10? | (Wei et al. 2005) |
CD8+/CD28-/FOXP3+ | Mouse | TERS-imprinted myeloid DC (arginase-dependent) | Impaired CTL afunction | NDb | (Mahadevan et al. 2012) |
18.7 The Effect of TERS In Vivo and Mechanism of Generation
Several lines of evidence suggest that TERS is operational in vivo. First C57BL/6 mice injected intra-peritoneally with TERS develop an ER stress response in liver cells characterized by the up-regulation of Grp78, Chop and spliced Xbp–1. This suggests that a tissue that is sensitive to ER stress induction, the liver, readily becomes a target of TERS administered systemically.
18.7.1 TERS-Imprinted Myeloid Cells Promote Tumor Progression In Vivo
The effect of TERS on tumor growth was examined in C57BL/6 mice inoculated subcutaneously with B16.F10 tumor cells admixed with TERS-imprinted bone marrow-derived DC according to Prehn (1972). Under these conditions, we noted an earlier tumor initiation, accelerated tumor growth, and decreased survival when compared to mice receiving B16.F10 tumor cells admixed with control bone marrow-derived DC, or tumor cells alone (Mahadevan et al. 2012). Thus, bone marrow-derived DC polarized by ER-stressed tumor cells facilitate tumor growth in vivo. B16.F10 tumors seeded with TERS-imprinted bone marrow-derived DC contained about half the percentage of tumor infiltrating CD8+ T cells as compared with control B16.F10 tumors. Interestingly, while we found a decreased number of CD8 T cells in tumors, we found no difference in the draining lymph nodes, implying the local nature of this phenomenon.
TERS-imprinted bone marrow-derived DC also function to dysregulate anti-tumor T cell immunity, allowing immune escape. For instance, TC1.OVA prostate cancer cells that constitutively express OVA, which functions as a tumor rejection antigen (Redmond et al. 2007), do not form tumors in mice reflecting their immunogenic status. However, when inoculated admixed with TERS-imprinted bone marrow-derived DC, they form transient tumors 6–10 days post-injection (Mahadevan et al. 2012).
18.7.2 TERS Is Produced In Vivo During Tumor Formation
New evidence shows that tumor-infiltrating myeloid cells in vivo display TERS characteristics. CD11b+ myeloid cells isolated from B16.F10 tumors implanted in C57BL/6 mice, or from spontaneous intestinal adenomas in adenomatous polyposis coli (APC) mice, display both an upregulation of the UPR and the mixed pro-inflammatory/suppressive phenotype typical of the TERS signature compared with bone marrow- or spleen-derived myeloid cells from tumor-bearing mice (Rodvold et al. 2014b).
18.7.3 TERS Requires Ire1α Signaling in Transmitter Cells
Because the ER stress response is under the control of three main sensors (IRE1α, PERK, ATF6), we began to deconvolute their precise role in the generation of TERS. Using MEFs each deficient in a single arm of the UPR, we found that compared with wild type MEFs the production of TERS was greatly diminished in IRE1α KO MEFs but not in PERK KO or ATF6 KO MEFs, providing the first indication that TERS generation may be mainly due to IRE1α signaling (Rodvold et al. 2014b). A hypothetical model of the signaling events involved in the generation of TERS is illustrated in Fig. 18.4.
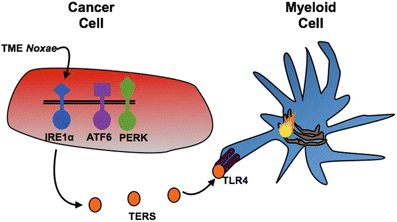
Fig. 18.4
Hypothetical model for the generation of TERS in transmitter cells. The model predicts that of the three main UPR sensors (PERK, ATF6 and IRE1α), Ire1α is responsible for the generation/release of the TERS factor(s) from stressed transmitter cancer cells. Myeloid cells (macrophages and dendritic cells) receive TERS factor(s), which is sensed in part by TLR4, and are polarized to a phenotype characterized by activation of the UPR (flame) and a pro-inflammatory/suppressive phenotype that facilitates tumor growth (see text for details)
18.8 Therapeutic Approaches Targeting the Tumor UPR
In the previous sections we discussed the cell-intrinsic role of the UPR in tumor adaptation and survival, as well as its putative cell-extrinsic role in polarizing myeloid antigen presenting cells to a phenotype that facilitates tumor outgrowth via T cell-dependent and independent mechanisms. Considering this dual role, targeting the UPR in the tumor microenvironment will likely have a dual benefit: impairing tumor cell microenvironmental adaptation and survival, and disabling a mechanism of host immune subversion. Based on our current understanding, the cellular targets, of any such intervention would be the tumor cell, myeloid antigen presenting cells, and CD8+ T cells (Fig. 18.5a). It remains to be seen whether CD4 T cell immunity is also adversely affected by the cell-extrinsic effects of the UPR.
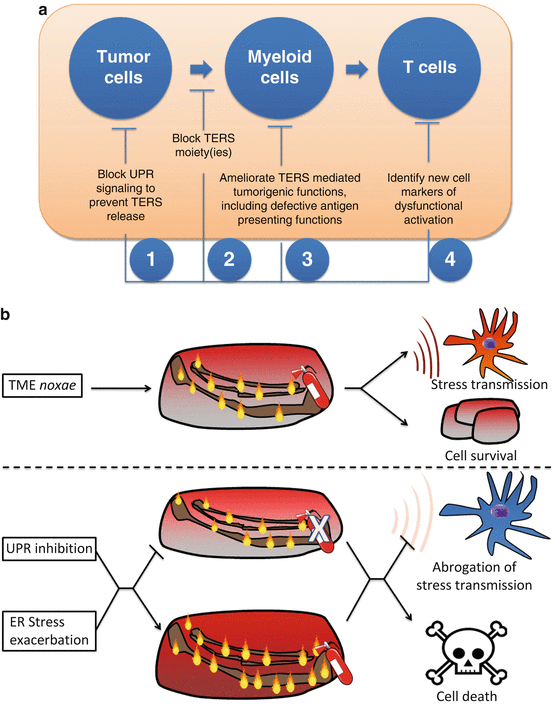
Fig. 18.5
Targeting the cell-extrinsic effects of the UPR in the tumor microenvironment. (a) Points of therapeutic intervention against the tumor-promoting effects of TERS. (b) Strategies for pharmacological control of the UPR in the context of cancer. (upper panel) Before any intervention, cancer cells exhibit a basal level of unfolded proteins (flames) compensated by the UPR (fire extinguisher). A combinatorial approach should involve tuning down the UPR (middle panel) with simultaneous exacerbation of ER stress (lower panel), hence inducing cell death in cells unable to cope with ER stress, as well as inhibiting the negative cell-extrinsic effects of the tumor UPR
The UPR is tumor microenvironment-specific as demonstrated by studies showing that peritumoral areas do not express UPR genes and that a constitutive UPR takes place within spontaneously growing tumors, though heterogeneously within a tumor mass (Spiotto et al. 2010). In addition, several lines of evidence indicate that UPR inhibitors selectively target tumor cells, as discussed below.
As the UPR represents an adaptive mechanism to cope with ER stress, targeting the UPR will likely take the following forms: (1) inhibition of the UPR in tumor cells with high levels of basal ER stress (eg. microenvironment-driven: hypoxia, glucose deprivation; tumor-intrinsic: secretory tumors, like myeloma), or (2) exacerbation of ER stress and consequent induction of cytotoxic/apoptotic effects. While each of these approaches will individually exploit tumor microenvironmental ER stress, either by its induction or by targeting its adaptive response (the UPR), they alone may not be sufficient to control the UPR within the complex and heterogeneous tumor microenvironment. For instance, exacerbating ER stress alone may exhibit cytotoxicity, especially in hypoxic/nutrient deprived areas; however, tumor cells mounting a UPR that leads to survival will have a UPR-mediated adaptive advantage, including resistance to chemotherapy (Pyrko et al. 2007) and host immunity. On the other hand, only inhibiting the UPR will target tumor cells with increased basal ER stress due to heterogenous microenvironmental noxae, sparing cells in more vascularized areas. We propose that optimal targeting of the UPR should take the form of inducing ER stress in tumor cells (fueling the fire) while concomitantly inhibiting the UPR (locking up the extinguisher) (Fig. 18.5b). In sum, this combinatorial mechanism will simultaneously take advantage of the cytotoxic potential of ER stress while inhibiting the response mechanism needed to adapt.
These strategies have already gained some experimental support. Bortezomib, a proteasome inhibitor that induces accumulation of proteins thus exacerbating ER stress, causes significantly higher cytotoxicity in hypoxic HeLa and human colorectal cancer cells than in normoxic cells, an effect dependent on ER protein load and consequent ER stress (Fels et al. 2008). Similarly, the induction of ER stress with a targeted thapsigargin pro-drug, celecoxib, or bortezomib, induces glioblastoma cell death, especially in hypoxic areas of the tumor (Johnson et al. 2002; Denmeade et al. 2012; Schonthal 2013). Combination of the ER stress inducers bortezomib and celecoxib, or its non-coxib analogue, 2,5-dimethyl-celecoxib (DMC), causes severe ER stress and apoptosis in murine glioblastoma cells in vitro and in vivo (Kardosh et al. 2008).
Inhibition of Xbp–1 splicing in multiple myeloma with the IRE1α endoribonuclease small molecule inhibitor, STF-083010, results in tumor cell-specific death in vitro and in vivo (Papandreou et al. 2011). Similarly, irestatin, a small molecule inhibitor of IRE1α endoribonuclease activity, inhibits hypoxic human myeloma and colon cancer cell survival and colony formation in vitro, as well as in vivo tumorigenesis in a heterotopic xenograft model (Papandreou et al. 2011). Targeting cell surface GRP78 in colon and lung cancer in mice with a monoclonal antibody (mAb159) causes tumor regression in vivo (Liu et al. 2013). Lastly, inhibition of GRP78 activation with active compounds present within the herbal medicine Ponciri fructis or the pyrone-type polyketide, verrucosidin, exhibits selective cytotoxicity in human pancreatic cancer cells or colon cancer cells undergoing glucose deprivation-induced ER stress (Park et al. 2007; Cha et al. 2009).
Inducing ER stress while concomitantly inhibiting the adaptive UPR has also begun to find experimental support. For instance, it has been shown that the mechanism of bortezomib’s cytotoxic activity in myeloma cells is its ability to inhibit Xbp1 splicing via stabilization of unspliced XBP-1, which acts as a dominant negative suppressor of XBP1-s, while inducing ER stress via proteasome inhibition (Lee et al. 2003a). Congruently, the induction of ER stress with bortezomib or 17-AAG in myeloma cells was shown to synergize with the activity of transgenic or small molecule-mediated inhibition of Xbp–1 splicing resulting in the induction of greater and irreparable cytotoxicity than either agent alone in vitro and in vivo (Lee et al. 2003a; Mimura et al. 2012). In human pancreatic cancer cells, bortezomib reduces GRP78 and CHOP expression under ER stress conditions and sensitizes them to ER stress-inducing compounds, including thapsigargin, tunicamycin, and cisplatin, yielding synergistic cytotoxicity in vitro and in vivo (Nawrocki et al. 2005). GSK2606414, a small molecule inhibitor of PERK autophophorylation and downstream eIF2α phosphorylation, cooperates with ER stress induced by hypoxia or thapsigargin, causing greater inhibition of in vitro clonogenic survival of pancreatic and colon cancer cells than either PERK inhibition or ER stress induction alone (Axten et al. 2012; Cojocari et al. 2013). Epigallocatechin gallate, which inhibits GRP78 by targeting its ATP-binding domain, sensitizes human glioma cells to ER stress induced by the chemotherapeutic agent, temozolomide, resulting in synergistic cyotoxicity, greater than either agent alone (Pyrko et al. 2007). There are several chemical UPR inhibitors that have displayed efficacy against tumor growth in vitro and in vivo (reviewed in (Li et al. 2011) and (Schonthal 2013)) these are presented in Tables 18.3 and 18.4.
Table 18.3
Tumor cell active ER stress/UPR modulators
Compound | Mechanism of action | Tumor cell type | Reference |
|---|---|---|---|
Thapsigargin, celecoxib | ER stress induction via SERCA inhibition | Breast, prostate | |
Biguanides, versipelostatin, pyruvinium pamoate | Inhibition of GRP78, ATF6, Xbp1 response to glucose deprivation | Fibrosarcoma, HeLa | (Saito et al. 2009) |
Verrucosidin, Epigallocatechin, Ponciri fructis active compound | Inhibition of GRP78 | Breast, glioblastoma | |
STF-083010, MKC-3946 | IRE1 endoribonuclease inhibition | Multiple myeloma | |
Irestatin | IRE1 endoribonuclease inhibition | Multiple myeloma, colon | (Papandreou et al. 2011) |
Bortezomib | IRE1 dominant negative inhibition | Multiple myeloma | |
ER stress induction via proteasome inhibition | |||
GSK2606414 | PERK kinase inhibition | Pancreas, colorectal |
Table 18.4
Cell-nonautonomous control of cellular stress responses
Stress response system | Organism | Transmitter (effectors) | Receiver (sensors) | Soluble mediators | Reference |
|---|---|---|---|---|---|
HSR | C. elegans | Thermosensory neuron (ND) | Muscle cell (ND) | ND (unc31-mediated) | (Prahlad and Morimoto 2011) |
HSR | C. elegans | Motor neuron (NT) | Muscle cell (postsynaptic NT receptor) | ACh, GABA | (Garcia et al. 2007) |
HSR | C. elegans | Muscle, intestinal cell (pha-4) | intestinal, pharyngeal, excretory cells (pha-4) | ND (unc31/unc13-independent) | (van Oosten-Hawle et al. 2013) |
UPRMT | C. elegans | Neuron (ND) | Intestinal cell (ND) | ND | (Durieux et al. 2011) |
UPRER | C. elegans | Neuron (ND) | Multiple somatic tissues (ND) | ND | (Sun et al. 2012) |
UPRER | C. elegans | Neuron (xbp1) | Intestinal cell (xbp1) | ND (unc13-mediated) | (Taylor and Dillin 2013) |
UPRER | Mouse | Epithelial tumor cell (IRE1) | Macrophage | ND | |
Human | Myeloid dendritic cell | ||||
Tumor cell (TLR4, IRE1) |
While there has been recent interest in developing UPR inhibitors active against tumor cells, there has been little or no investigation the effect of tumor UPR inhibition on the host anti-tumor immune response. A link between the tumor UPR and the immune response was originally suggested by the finding that silencing of Grp78 in mouse fibrosarcoma cells inhibited growth in an in vivo syngeneic transplantation model due, in part, to increased tumor cell-specific memory T cell generation (Jamora et al. 1996). In addition, overexpression of GRP78 in murine insulinoma cells leads to impaired CD8 T cell priming and inhibition of killing, when GRP78-overexpressing tumor cells were used to prime cytotoxic T cell lines, as targets, respectively (Wang et al. 2007). Discovery and characterization of the effect of TERS on host immunity has continued this line of inquiry (Zanetti 2013).
Based on our findings on transmissible ER stress, it appears that a fruitful avenue for therapeutic development will be to develop decoy systems (antibodies, aptamers, etc.) to intercept the TERS factor(s) in the extracellular space (Fig. 18.5a). In this scenario neutralization of TERS would also inhibit the polarization of myeloid cells to a pro-inflammatory/suppressive phenotype, and in turn prevent and unfetter the untoward effects on T cell-mediated immunity, perhaps permitting more effective autochthonous or vaccine-induced anti-tumor immune responses. In addition, TERS may induce tumor-infiltrating myeloid cells to produce tumorigenic cytokines and adversely affects antigen presentation (see Sects. 3.2 and 3.3 for discussion). Lastly, as the downstream effects of TERS on T cell priming are elucidated (i.e. polarization toward a suppressive phenotype), new targets for therapy will come to light (e.g., ecto-ATPases, immune checkpoint molecules, UPR signaling components). Targeting the tumor-infiltrating myeloid cell UPR, the tumor cell UPR, and ultimately suppressive T cells, will reset the multifaceted dysregulation of the tumor microenvironment that hinders anti-tumor immunity.
18.9 Conclusions and Perspectives
During their growth, tumor cells are subjected to noxae that exist in the tumor microenvironment and are able to induce the ER stress response that, as discussed, leads to multifaceted dyregulation. Some of them are cell-intrinsic in nature and promote tumor cell adaptation and survival. Others are cell-extrinsic and affect the function of neighboring cells—immune cells, cancer cell themselves (Rodvold et al. 2014a), and stromal cells in a significant way. The intent of this chapter was to bring attention to effects on immune cells.
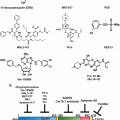
A hitherto unappreciated phenomenon, transmissible ER stress (TERS), seems to link together tumor cells and immune cells. Receiver myeloid cells, macrophages and dendritic cells become polarized, via ER stress transmission, to a mixed, pro-inflammatory/suppressive phenotype. The pro-inflammatory component, an exquisite innate immune trait, is per se sufficient to fuel tumor growth through a variety of mechanisms including perhaps increasing tumor cell proliferation, survival, and the mutational rate (Grivennikov et al. 2010; Tili et al. 2011). Importantly, however, we found that a mixed, pro-inflammatory/suppressive phenotype also hampers essential immunological functions of dendritic cells, i.e., antigen presentation and T cell priming, hence disabling the adaptive T cell response (Mahadevan and Zanetti 2011; Zanetti 2013). Combined, these effects favor faster tumor growth (Fig. 18.6).
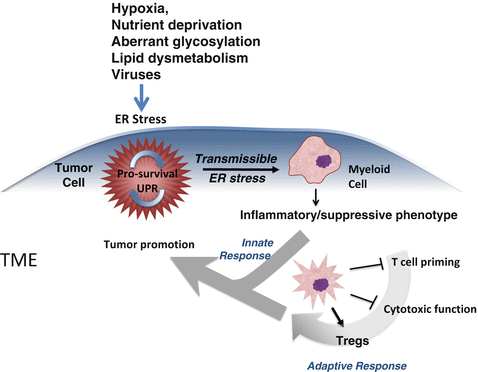
Fig. 18.6
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities




Cell non-autonomous regulation of immunity within the tumor microenvironment via transmissible ER stress. Various noxae in the tumor microenvironment (TME) perturb tumor cells inducing a UPR. Under the umbrella of the UPR response (blue shade) there occur multiple events, which ultimately influence neighboring myeloid cells and T cells. In this framework, the tumor UPR acts as a cell-intrinsic tumor pro-survival factor (circular arrow). ER-stressed tumor cells also release many factors (cytokines, chemokines metalloproteinases etc.) among which is a factor(s) able to polarize myeloid cells by transmitting ER stress to them (second hand stress). As a result receiver myeloid cells undergo a UPR, and acquire an inflammatory/suppressive phenotype that facilitates tumor growth directly (innate response) and/or via dysregulation of T cell immunity (adaptive response). Together, these T cell-independent and –dependent effects (merging arrows) favor tumor growth (Adapted from Mahadevan and Zanetti (2011))
Related posts:
Introduction to Cell Stress Responses in Cancer: The Big Picture
Pro-senescence Therapy for Cancer: Time for the Clinic
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
