Fig. 3.1
The cell cycle and its checkpoints. Cyclins are individually synthesised and degraded in a phase distinctive manner and bind and activate specific Cdks which coordinates the sequential progression of the cell cycle phases. Four main checkpoints, one in each cell cycle phase, respond to different types of DNA insults and promote cell cycle arrest and DNA damage repair
3.2.2 Cell Cycle Checkpoints
The cell division cycle is surveyed by multiple checkpoints that respond to a wide range of internal and external stresses to ensure genomic integrity from one cell generation to the next. When checkpoints are triggered, CDKs are generally inhibited (the exception being the spindle assembly checkpoint in mitosis where inactivation of the CDK is inhibited); this results in cell cycle arrest and provides time for DNA repair to occur. If the damage is extensive and cannot be resolved, checkpoints trigger cell senescence or apoptosis.
There are four main checkpoints responding to different types of DNA insults, one in each cell cycle phase (Fig. 3.1). The G1 phase checkpoint prevents the replication of damaged genomic material by blocking entry into S phase, the S phase checkpoint not only responds to the presence of DNA damage but also to aberrant replication forks by stopping or slowing DNA synthesis, the G2 phase checkpoint impedes cells with damaged or entangled, catenated DNA from undergoing mitosis, and finally the spindle assembly checkpoint only allows mitotic exit if the chromosomes are properly attached to the mitotic spindle.
3.2.3 Cell Cycle Signalling Pathways
Two major players in checkpoint signalling in response to DNA damage are the ataxia telangiectasia mutated (ATM) and the ataxia telangiectasia and Rad3-related protein (ATR) proteins. ATM is mainly triggered by double strand DNA breaks (DSBs), while ATR primarily responds to single-stranded DNA (ssDNA) (Fig. 3.2). Once activated, these kinases phosphorylate and activate checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2) proteins, which in turn target a range of effectors that inhibit cell cycle progression, including the immediate activators of the CDK/Cyclins, the Cdc25 phosphatases. ATM/ATR and Chk2/Chk1 also phosphorylate and activate p53 protein which promotes the transcription of p21, GADD45 and 14-3-3 CDK inhibitor proteins (Elledge 1996; Ciocca and Elledge 2000; Elledge et al. 2000; Huang and Elledge 2000; Liu et al. 2000; Schulman et al. 2000; Tibbetts et al. 2000; Wang et al. 2000; Zhou and Elledge 2000), thus enhancing cell cycle arrest. Another mitotic regulatory pathway involves Plk1 protein kinase which is activated by the cooperative action of Aurora A protein kinase and its cofactor Bora (Seki et al. 2008; Takaki et al. 2008). Plk1 activity is also regulated by ATM/ATR checkpoint signalling (Smits et al. 2000; van Vugt et al. 2001). A link between this pathway and the G2 phase DNA damage checkpoint has been recently described where Bora is degraded via ATR-mediated phosphorylation following DNA damage (Qin et al. 2013), and ATM-dependent phosphorylation and activation of B56 regulatory subunit of PP2A that is responsible for dephosphorylating and inactivating Plk1 (Shouse et al. 2011).
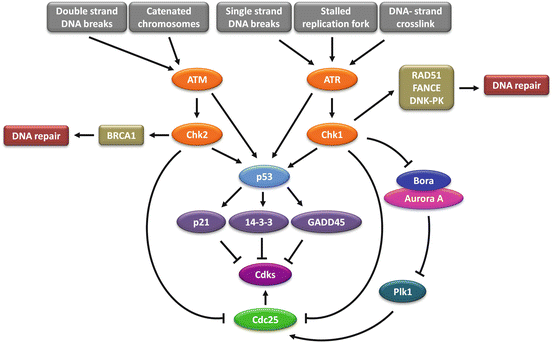
Fig. 3.2
Cell cycle checkpoints signalling pathways. In response to different types of DNA damage, ATM and ATR are activated and subsequently upregulate Chk1 and Chk2. In turn, Chk1 and Chk2 inhibit Cdks by deactivating Cdc25 and by promoting the transcription of p21, 14-3-3 and GADD45 Cdks inhibitory factors via p53 activity. Differently from all the others, the spindle assembly checkpoint maintains Cdk1/Cyclin B activity which in turn hampers mitotic progression. Beyond promoting a cell cycle checkpoint arrest, Chk1 and Chk2 also activate the DNA damage repair mediated through RAD51, FANCE, DNK-PK and BRCA1 proteins
In response to DNA lesions, cells initiate a highly coordinated cascade of events, known as the DNA damage response (DDR), which is essential for the maintenance of genomic stability and cell survival. DNA damage can interfere with essential cellular processes, such as transcription or replication, and can compromise the viability of the cells. DNA damage repair is tightly coordinated with cell cycle progression through the activation of DNA damage checkpoints. The DDR consists of detection of damage by sensor proteins, with signal transducer and effector proteins launching a cascade of events that causes cell cycle arrest, activation of DNA repair, senescence or apoptosis.
DNA damage is detected by sensor proteins, with DSBs detected by the MRE11-RAD50-NBS1 (MRN) sensor complex and ssDNA arising from DNA damage processing is signalled by Replication protein A (RPA), an ssDNA binding protein. Once DNA damage is sensed, the cell must transduce this signal down to its appropriate effectors. The MRN complex leads to the recruitment and activation of ATM and RPA recruits the ATR kinase via its partner protein, ATRIP (ATR-interacting partner) (reviewed in Elledge 1996; Zhou and Elledge 2000). ATM and ATR are related kinases that phosphorylate a number of mediators which are mostly cell cycle specific and associate with damage sensors, signal transducers and effectors at particular phases of the cell cycle, and as a consequence, provide signal transduction specificity. ATM and ATR are both members of the PI-3K-like kinase family (PIKK) of protein kinases that also includes DNA dependent protein kinase (DNA-PK) and mammalian target of rapamaycin (mTOR). The ATM signalling cascade includes 53BP1, MDC1, BRCA1, MCPH1, and PTIP, while mediators of ATR signalling include TopBP1, and Claspin (reviewed in Marechal and Zou 2013). The transducer kinases lead to the activation of the effector kinases CHK1 and CHK2, activating signalling cascades with downstream targets including transcription factors, cell cycle regulators, apoptosis and DNA repair factors (Fig. 3.2). There is crosstalk between the ATM/Chk2 and ATR/Chk1 pathways and they share many substrates.
3.3 DNA Damage Response and Repair Mechanisms
It is estimated that each of the ~1013 cells within the human body incurs tens of thousands of DNA-damaging events per day (Lindahl and Barnes 2000). To avoid the deleterious consequences of damage accumulation, multiple DNA repair pathways have evolved, each associated with specific classes of lesions.
DNA is subject to a high level of endogenous damage resulting from reactive oxygen species (ROS) generated from normal cellular metabolism, in addition to a number of other factors including spontaneous hydrolysis, abasic sites, and alkylation. Endogenous sources of damage can arise from physiological DNA processing and DNA repair processes themselves, such as DNA mismatches, insertions and deletions which can be introduced as a result of misincorporation of bases by replicative DNA polymerases (McCulloch and Kunkel 2008).
There are a number of exogenous or environmental sources of DNA-damaging agents including ultraviolet radiation (UV), ionizing radiation (IR), chemical agents used as clinical and chemotherapeutic drugs, tobacco smoke and other environmental agents. Environmental stresses such as ultraviolet light (UV) from the sun, primarily causes two types of DNA lesions, cyclobutane pyrimidine dimers (CPDs) and 6–4 pyrimidine products (6–4PP) (You et al. 2001). IR is the most common DSB-inducing agent, and can originate from both natural (gamma and cosmic radiation) and artificial sources (eg. X-rays and radiotherapy). IR can also induce DNA damage indirectly through the production of ROS.
Chemical agents used as chemotherapeutic drugs, including alkylating agents (eg. temozolomide), bifunctional alkylating agents (e.g. platinum agents), and mitomycin C cause DNA damage in the form of intra-strand and inter-strand cross-links. Targeted drugs such as topoisomerase I or II inhibitors, generate ssDNA or dsDNA breaks by trapping topoisomerase-DNA covalent complexes (Sinha 1995). Nitrosamines are a class of potential carcinogens that are found in tobacco smoke, and there is a possible dose-response relationship between the amount and duration of tobacco smoke exposure and mutational burden (Govindan et al. 2012). Other naturally occurring environmental agents include N-nitrosoamines, heterocyclic amines, and polycyclic aromatic hydrocarbons which are common in the diet, producing bulky DNA adducts (reviewed in Dexheimer 2013).
3.3.1 Types of DNA Damage Repair
DNA damage repair can be subdivided into several distinct mechanisms based on the type of DNA lesion (Fig. 3.3), summarised below.
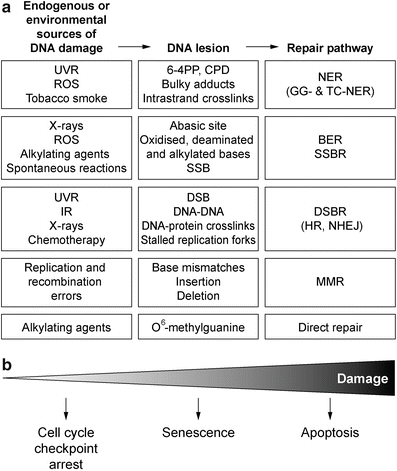
Fig. 3.3
DNA damage repair. (a) Sources of DNA damage and the specific mechanisms used to repair the DNA lesions. (b) The extent of DNA damage and success of repair determine whether the cell undergoes cell cycle checkpoint arrest to allow for repair, or where damage is too extensive the cell encounters senescence, or death
3.3.1.1 Nucleotide Excision Repair (NER)
NER is the major repair pathway for removal of bulky DNA adducts which distort the normal structure of the DNA helix. These can be formed by ultraviolet radiation (yielding adducts like thymidine dimers or 6–4 photoproducts), chemicals or ROS, which produce lesions that disrupt transcription and replication. NER also contributes to the repair of intra- and interstrand crosslinks (ICLs). Two NER sub-pathways exist: global genome NER (GG-NER) which repairs damage that occurs throughout the entire genome, while transcription-coupled repair (TC-NER) preferentially repairs damage in transcriptionally active genes (reviewed in Naegeli 1995). Both pathways converge into a common pathway involving over 20 different genes. Hereditary defects in NER cause UV sensitivity and skin cancer development (Andressoo et al. 2005). There are three separate though inter-related autosomal recessive disorders including Xeroderma pigmentosum, Cockayne Syndrome and Trichothiodystrophy (TTD). Subjects with Xeroderma pigmentosum exhibit >100-fold increased incidence of sun-induced skin cancer (Arlett and Lehmann 1996).
3.3.1.2 Base Excision Repair (BER)
BER is involved in the removal of a variety of endogenous and exogenous DNA lesions, repairing mostly alkylated, deaminated and oxidized bases, in addition to abasic sites. BER is a multistep process that requires the sequential activity of more than 20 proteins and consists of several distinct BER sub-pathways, dependent on the type of DNA damage encountered, with different glycosylases detecting different types of base damage (Dizdaroglu 2005). Mutations and polymorphisms in BER genes have been associated with aging, cancer susceptibility and neurodegeneration (Dianov and Hubscher 2013).
Single Strand Break Repair (SSBR)
Single strand breaks (SSBs) in DNA are the most common endogenous lesions, arising both directly from DNA damage induced from ROS and indirectly from BER (reviewed in Caldecott 2008). Whilst SSBs are not as harmful as DSBs, they can block DNA replication and transcription, and occur three orders of magnitude more frequently than DSBs (Bradley and Kohn 1979). SSBs can arise as BER intermediates, there are also numerous involuntary ssDNA breaks that can also occur through direct and indirect mechanisms. Exogenous sources of SSBs include IR and UV radiation that can generate SSBs during lesion repair, or can be the result of genotoxic agents such as chemotherapeutic drugs. The most likely consequence of unrepaired SSBs is the blockage or collapse of DNA replication forks during S phase, possibly leading to the formation of DSBs.
Repair of SSBs occurs by a GG-NER pathway throughout the genome and throughout the cell cycle to rapidly detect and remove the majority of chromosomal SSBs (reviewed in de Laat et al. 1999). However, SSBs encountered by the DNA replication machinery during S-phase can cause the replication fork to stall, risking replication fork collapse. Cells have developed a DNA-damage tolerance pathway known as post-replication repair, which enables the cells to complete bulk DNA replication and remove the lesions after replication is completed (Wigan et al. 2012). This may be undertaken using one of two mechanisms, an error-free recombination repair mechanism, and an error-prone translesion synthesis (TLS) mechanism. Defects in SSB repair cause neurological disorders including Ataxia-oculomotor apraxia 1 (AOA1), and Spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) (Rass et al. 2007).
Double Strand Break Repair (DSBR)
DNA DSBs are the most deleterious form of DNA damage because they do not leave an intact complementary strand to be used as a template for DNA repair. If left unrepaired, they can ultimately lead to chromosome breaks and translocations (Jackson and Bartek 2009). Hence repair of DSBs is both critical for cell survival and maintenance of genome integrity. DSBs can be generated in response to IR, many chemotherapeutics (etoposide, doxorubicin, camptothecin derivatives) which act as topoisomerase poisons, from replication of DNA containing SSBs, and naturally occurring DSBs arising at chromosome ends when telomeres become critically short during replicative senescence. DSB repair utilises two major pathways: homologous recombination (HR) and non-homologous end-joining (NHEJ). These pathways are complementary and operate optimally under different circumstances.
Homologous recombination is a highly complex process that involves multiple proteins and occurs during late S and G2 phases of the cell cycle, requiring the presence of a homologous template (for review see Li and Heyer 2008). The broken DNA ends of a DSB are resected to allow invasion of the single strands into the sister chromatid, which functions as a template for accurate resynthesis of the damaged DNA. This pathway repairs only a minor proportion of DSBs, however it may be the most crucial as it is high fidelity, and it deals with stalled and collapsed replication forks, as well as single-ended DSBs, and also ICLs. HR is crucial for the maintenance of genomic stability, and the function of the entire pathway can be compromised by mutation in one or more genes. Many tumour suppressor genes participate in this pathway including BRCA1 and BRCA2 which are mutated in hereditary breast cancers, and ATM.
NHEJ repairs double-stranded DNA breaks without the need for template DNA. NHEJ is active in all phases of the cell cycle, predominating in G1 phase, and mediates the direct ligation of broken DNA ends, with minimal DNA end processing. NHEJ is the prevalent DSB repair pathway, however it has the potential to be error prone, as deletions or insertions can be induced at sites of repair (Lieber et al. 2010). While there have been reports of some single nucleotide polymorphisms in NHEJ associated gene loci linked to cancer and epigenetic silencing of NHEJ components, defects in members of the NHEJ pathway have infrequently been identified in association with cancer. Cells that lack any of the NHEJ components are radiation sensitive.
3.3.1.3 Mismatch Repair (MMR)
The MMR pathway repairs errors that have occurred during normal DNA replication caused by the incorporation of the wrong nucleotide, nucleotide deletions or insertions, and have escaped the proofreading activity of replicative polymerases. MMR is also required for the removal of bases damaged by methylating agents and antimetabolites and possibly intrastrand crosslinking agents (chemotherapy). There are essentially three steps in the repair process: a recognition step where mispaired bases are recognized, an excision step where the error-containing strand is degraded resulting in a gap, and a repair synthesis step, where the gap is filled by the DNA resynthesis (for review of MMR, see Stojic et al. 2004).
Defects in MMR leads to a mutator phenotype which causes cancer predisposition and also affects DNA damage signaling, recombination, and several other DNA metabolic processes (Jiricny 2006). Mutations in MMR genes are associated with hereditary non-polyposis colon cancer (HNPCC), with MMR defective cells unable to correct errors caused by DNA polymerase slippage at repetitive sequences in the genome (for review see Cleaver et al. 2009).
3.3.1.4 Oxidative Damage Repair
O6-methylguanine-DNA methyltransferase (MGMT) demethylates O6-methylguanine lesions, which are formed as a result of erroneous methylation and other alkylations induced by dietary nitrosamines or chemotherapy agents. Unrepaired O6-methylguanine is mutagenic because distorted pairing with cytosine or thymidine leads to G:C to A:T transitions on replication (Saffhill et al. 1985).
3.4 Defective Cell Cycle Checkpoints and DNA Damage Repair in Cancer
Defective cell cycle checkpoint function may increase spontaneous and induced gene mutations and chromosomal aberrations by reducing the efficiency of DNA repair. Cell cycle checkpoints integrate DNA repair with cell cycle progression and are commonly deregulated in cancers leading to increased genomic instability. This provides cancer cells with an evolutionary or adaptive advantage, allowing them to modify their transcriptome and/or genome to increase their ability to thrive in new tissue environments.
Cell cycle checkpoint pathways integrate repair of specific DNA lesions with the cell cycle position. The phase of the cell cycle in which a checkpoint is induced, and the duration of the arrest is dependent on the type of triggering DNA lesion, the number of lesions and the length of time required to repair the lesions (for review see Lazzaro et al. 2009). There are some lesions that are rapidly repaired and do not require cell cycle checkpoint arrest, while in some cases the cells may keep cycling and reach a cell cycle phase where the specific damage is less toxic or more easily repaired.
G1/S phase transition is a major target of alterations in cancer. Such alterations often result in changing the Retinoblastoma protein (Rb) phosphorylation/dephosphorylation balance and consequently effecting the cell proliferation and blocking entry into S phase (Hall and Peters 1996). Rb point mutations or deletions are found in a wide variety of human cancers, particularly retinoblastomas and sarcomas. Histone deacetylase (HDAC)-Rb complex formation is also inhibited by HDAC point mutations found in human cancer (Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998).
P16-mediated senescence is another G1 phase mechanism that responds to extensive DNA damage (Robles and Adami 1998; Shapiro 2006). CDKN2A is the gene which encodes the cell cycle inhibitor p16 INK4A , which is commonly defective in melanoma (Bartkova et al. 1996; Castellano et al. 1997; Hayward 2003). P16, a major driver of senescence, creates G1 phase arrest by blocking Rb inactivation through CDK4/6-Cyclin D inhibition. Melanoma associated mutation of p16 disrupts its ability to promote senescence arrest (Haferkamp et al. 2008). In addition to this role, increased p16 expression has been correlated with the G2 phase checkpoint arrest in response to suberythemal UVR (Pavey et al. 1999, 2001; Abd Elmageed et al. 2009).
The tumour suppressor p53 is involved in DNA damage response and is inactivated in 50 % of all human cancers (Hollstein et al. 1991), and humans carrying a germ-line deletion of one TP53 allele are highly prone to cancer development (Malkin et al. 1990; Srivastava et al. 1990). In the face of a range of genotoxic insults, p53 is rapidly stabilized and its increased level promotes cell cycle arrests in G1 and G2 phases in cell lines in vitro and signals apoptosis in response to excessive damage. The G1 checkpoint is activated by ATM signalling to Chk2 and p53, among other targets, preventing the replication of damaged DNA by blocking entry into S-phase. ATM promotes Homologous recombination repair by recruiting BRCA1 to DSBs, but can also antagonize BRCA1 and promote NHEJ by recruiting 53BP1. Defects in the function of p53 may contribute to the genetic instability that appears to drive neoplastic evolution.
The decatenation checkpoint is another G2 phase checkpoint that has been reported to be defective in a large proportion (68 %) of melanoma cell-lines (Brooks et al. 2014). This ATM/Chk2-dependent checkpoint ensures that chromosome catenation, normally resulting from DNA replication, is resolved before the cell progresses into mitosis. The difference in checkpoint functional response was not attributable to Topoisomerase II levels. Intriguingly, despite the absence of cycle arrest, the checkpoint defective cell-lines were able to activate the checkpoint signalling following treatment with the topoisomerase II inhibitor ICRF-193, indicating participation of other players, possibly involved in checkpoint recovery, in this phenomenon (Brooks et al. 2014).
The mitotic spindle assembly checkpoint (SAC) monitors spindle defects. Many of the spindle assembly checkpoint sensors and effectors are localized to kinetochores, including the Mad and Bub gene families and Aurora kinases. This checkpoint detects defects in spindle attachment to the replicated chromosomes through the centromere localised kinetochore complex, and tension across the centromere, to ensure the fidelity of partitioning of the replicated genome into the daughter cells. Although mutations in SAC genes are uncommon, they have been reported at low frequency in a number of cancer types (Cahill et al. 1998). The microtubule direct drugs, taxane, vincalkaloids, and some of the newer targeted drugs such as Aurora inhibitors target this checkpoint. Cancer cells appear less able to maintain the mitotic arrest in the face of these drugs resulting in mitotic slippage and triggering cell death (Dowling et al. 2005; Weaver and Cleveland 2005; Gabrielli et al. 2007; Stevens et al. 2008).
3.5 Exploiting Defective Cell Cycle Checkpoints and DNA Damage Response in Cancer Therapy
Increased endogenous damage levels in cancer cells caused by defects in the cell cycle checkpoints and deregulation of DNA repair mechanisms can lead to genomic instability and contribute to the initiation and progression of cancer. This must be accommodated by the cells employing either novel mechanisms to cope with the stress, an adaptation to accommodate the stress, becoming more reliant on alternative stress response mechanisms or a combination of all three. Up-regulated DDR pathways can confer therapy resistance to DNA-damaging chemotherapy and radiotherapy, while down-regulated DDR pathways can lead to dependence on a compensatory pathway. This increased reliance on alternative mechanisms to cope with a stress makes it plausible to disarm these mechanisms to selectively target the cancer cells population while normal cells are being protected by their intact checkpoint and repair responses.
3.5.1 Chemotherapy
Most of the conventional chemotherapeutic drugs target rapidly proliferating cell populations by creating high levels of DNA damage and causing cell cycle arrest and cell death. Despite their broad clinical applications, there are serious drawbacks to the use of DNA damaging agents such as a narrow therapeutic index due to poor selectivity for cancer cells compared to normal proliferating cells, and a frequent loss of effectiveness due to innate or acquired resistance to chemotherapy. Resistance to the treatment can emerge from multiple mechanisms including drug uptake and metabolism, alterations in DNA repair pathways and defects in apoptotic mechanisms (Fodale et al. 2011). Cells respond to DNA damage and/or aberrant replication stress by activating cell cycle checkpoints, slowing cell cycle progression in order to facilitate DNA repair or promoting cell death with irreparable DNA lesions (Langerak and Russell 2011). As a result, sensitivity to these chemotherapeutic agents is reduced by normal checkpoint function.
3.5.1.1 Alkylating Agents
Alkylating agents are one of the common types of chemotherapeutic drugs that prevent the cancer cells from proliferating by creating intra- and interstrand crosslinks in DNA. The alkylating agents are not phase-specific and work in all phases of the cell cycle. Depending on the p53 status, they will cause G1, S and G2 phase arrests in cancer cells. In p53 wild type cells, p53 dependent activation of DNA damage repair components will promote repair and result in diminished activity of alkylating agents (Barckhausen et al. 2014). For instance, Dacarbazine (5-(3,3-Dimethyl-1-triazenyl) imidazole-4-carboxamide, DTIC) only displays 10–20 % response rates with no demonstrated impact on overall survival in melanoma patients (Chapman et al. 1999; Wagner et al. 2000), while temozolomide, an orally available imidazotetrazine derivative of dacarbazine, also demonstrates the similar pattern (Middleton et al. 2000). Defects in Mismatch Repair (MMR) cause tolerance to temozolmide, platinum agents and some nucleoside analogues, leading to drug resistance, with adverse reactions observed in clinical trials (reviewed in Kinsella 2009).
3.5.1.2 Antimetabolites
Antimetabolites, such as 5-FU and gemcitabine, deter DNA synthesis of cancer cells by inhibiting either thymidylate synthase or ribonucleotide reductase and, in addition, they disrupt strand synthesis by incorporating into DNA (Iwasaki et al. 1997; Longley et al. 2003). Thus, these antimetabolites can trigger an S phase arrest and reduce the efficacy of these drugs through the normal checkpoint role of blocking cell cycle progression and facilitating repair of the drug induced damage. For example, as high as 50 % of metastatic colorectal cancer patients are insensitive to 5-FU-based chemotherapy (Douillard et al. 2000; Giacchetti et al. 2000; Alhopuro et al. 2005). Gemcitabine, which is now considered as the standard treatment, has a response rate of less than 20 %, although it does provide an improvement in the quality of life in pancreatic cancer patients (Burris et al. 1997).
Genotoxic therapies trigger ATM-/ATR-dependent checkpoint arrest and the checkpoint response defects dependent on these signalling pathways indicate that differentiating patients on the basis of checkpoint signalling or repair defects in their tumours could increase the response and survival rates. For instance, in breast cancer patients, BRCA mutant patients defective for homologous recombination repair of the cisplatin induced DNA damage responded better to cisplatin treatment (Silver et al. 2010; Curtin 2012). Defects in NER have been used to confer sensitivity to platinum based chemotherapeutics, which reflects a reduced capacity to repair ICLs (Usanova et al. 2010). Alterations in the XPC gene (ERCC5) expression through loss of heterozygosity (LOH) and polymorphisms have been shown to be prognostic indicators to platinum based chemotherapy in lung, ovarian and colon cancers.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


