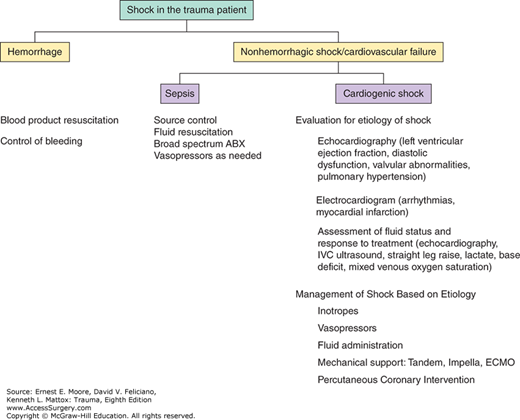
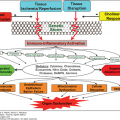
Cardiovascular failure is deterioration of cardiac function or vascular tone that results in impairment in end-organ perfusion. Collapse of the cardiovascular system can be either the end result of multisystem organ failure (MOF) or a manifestation of impaired cardiac function. In the acute phase of trauma, most shock is due to hemorrhage. Hemorrhagic shock leads to decreased preload or decreased right heart filling volumes, which is compensated by tachycardia and then may progress to hypotension and end-organ hypoperfusion. Cardiogenic shock alone stems from impaired myocardial contractility resulting in impairment of end organ perfusion with elevated cardiac filling pressures and low cardiac output.1 Unlike hemorrhagic shock, cardiogenic shock often does not respond simply to volume/blood product resuscitation and control of hemorrhage.2,3 Cardiogenic shock solely from impaired contractility is most often due to an acute myocardial infarction or acute on chronic heart failure.4 Therapy to support the failing cardiovascular system is directed at the etiology of the shock state and includes fluid resuscitation (preload) as well as pharmacologic modulation of vascular tone (afterload), contractility (with inotropes), and heart rate (with chronotropes). This chapter will explain the physiologic components of cardiovascular failure, assessment of patients with cardiovascular failure, treatment and monitoring (Fig. 56-1).5
Cardiac output is defined as the quantity of blood ejected into the aorta by the heart each minute and is calculated as heart rate multiplied by stroke volume (CO = HR × SV). This is the quantity of blood that flows through the circulation and is responsible for oxygen and nutrient transport to the tissues. The primary determinants of cardiac output are preload (the venous return to the heart), afterload (the resistance against which the heart must pump), contractility (the extent to which the myocardial cells can contract), and heart rate. The primary determinant of cardiac output is the filling of the heart and the ability to pump that volume effectively. Accordingly, the majority of therapies for augmenting cardiac output aim to restore filling pressures and support ineffective contractility.
Typically, 5.6 L/min is considered a normal resting cardiac output as measured in young, healthy males. However, cardiac output varies with activity, and is influenced by level of metabolism, exercise state, age, size of the individual, and other factors. Cardiac output in women is generally stated as being 10–20% lower than in men. Additionally, when factoring in age, the average cardiac output for adults is approximated as 5 L/min. Laboratory and clinical research have demonstrated that cardiac output increases in proportion to increasing body surface area. Therefore, to standardize cardiac output measurements between individuals, cardiac index (defined as cardiac output divided by body surface area in m2) is employed.6
Preload is the force that stretches the myocardium prior to contraction and most commonly is fluid dependent. The Frank–Starling relationship shows that as cardiac sarcomere length increases so will the force of contraction, until the sarcomere is stretched beyond its maximal length at which time the force decreases. The Frank–Starling relationship provides a paradigm by which the cardiovascular system and its derangements can be approached. Hypovolemia, or decreased preload, is the result of hemorrhage, contraction of the intravascular space due to external fluid losses, or contraction of the intravascular space due to internal sequestration as edema or third-space losses. Taken together, intravascular volume and venous return determine left ventricular end-diastolic volume (LVEDV), which in turn determines the force of ventricular contraction. The pulmonary artery catheter (PAC) is used to measure the pulmonary arterial wedge pressure (PAWP), which approximates left ventricular end-diastolic pressure (LVEDP). Assuming unaltered ventricular compliance, LVEDP theoretically approximates the LVEDV. Unfortunately, in the setting of critically ill patients, factors such as myocardial ischemia, heart failure, and myocardial edema can decrease ventricular compliance, rendering measurement of PAWP as a surrogate for LVEDV unreliable. In these situations, normal or elevated PAWP may not eliminate inadequate preload as a cause of low cardiac output. Therefore, multiple measures of cardiovascular function including the pulmonary artery pressure, CVP, IVC filling, clinical scenario, and response to fluid challenge all should be used to gauge preload when caring for these complex patients.
Besides changes in venous capacitance, venous return to the heart can be compromised by increased intra-thoracic or intra-abdominal pressure. This is most evident with tension pneumothorax, when the shock state is immediately reversed by decompression of the pleural space. Decreased venous return is also seen with abdominal compartment syndrome and pregnancy, of which the former can lead to dramatic hypotension following intra-abdominal trauma.
Cardiovascular failure due to a reduction in afterload is referred to as distributive shock, and has multiple etiologies: sepsis, neurologic deficit, and anaphylaxis. In trauma patients, distributive shock may develop from sepsis later in the post-trauma recovery period and is unlikely to be the source of shock in the immediate post-trauma period unless there is severe CNS injury. Afterload is the force that opposes ventricular contraction. Afterload is estimated with systemic vascular resistance. SVR is calculated using the hemodynamic equivalent of Ohm’s law:
In this equation, MAP is the mean arterial blood pressure, CVP is the central venous pressure, and CO is the cardiac output. This equation provides an approximation of vascular impedance. Therefore, it is important to realize that the clinical practice of “measuring” afterload or SVR with data from the PAC actually provides a calculated value that assumes nonpulsatile flow and does not consider the viscosity of blood, the elastic properties of the arterial walls, or the changes in microvascular resistance. Finally, because SVR is inversely proportional to cardiac output, rather than directly treating an abnormally high SVR, one should first treat the low CO with fluid administration to maximize preload, which will serve to increase CO and decrease SVR.
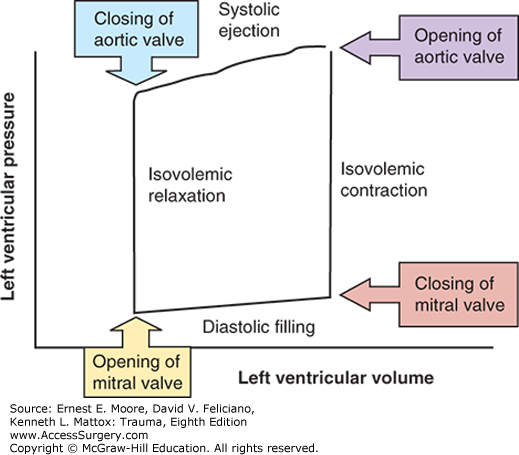
Contractility, also known as inotropy, is the force with which the myocardium contracts. The inotropic state of the myocardium, and the stroke work performed, can be visualized by the construction of a left ventricular pressure–volume loop (Fig. 56-2). This loop is bounded by the four phases of the cardiac cycle: isovolemic relaxation, diastolic filling, isovolemic contraction, and systolic ejection. Stroke work is defined as the area bounded by this loop.
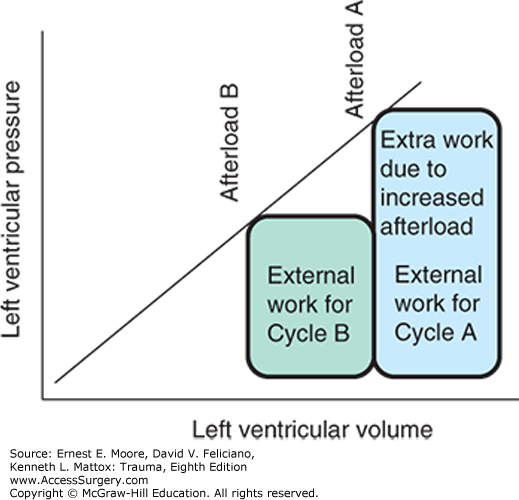
Instantaneous pressure–volume curves also provide a method to determine both external (stroke work) and internal (loss as heat) work performed by the heart during the cardiac cycle. As described above, the area bounded by the pressure–volume loop defines the external, or stroke work, performed by the myocardium. The internal work is defined as the area of the triangle determined graphically by three lines: an extrapolation of the elastance line to the x-intercept, the isovolemic relaxation portion of the pressure–volume loop, and the diastolic filling portion of the pressure–volume loop extrapolated back to its x-intercept (Fig. 56-3). Under this system, the failing heart with low contractility will demonstrate a shallow elastance line, which translates to low efficiency (more internal work performed for the same external work). Clinically, this points toward manipulation of contractility to balance myocardial oxygen demand and delivery. Finally, the strong influence of changes in afterload is readily appreciated under this model, as increasing afterload at a given stroke volume results in increased external work performed by the heart.
Heart rate is a key determinant of cardiac output. In the setting of constant stroke volume, increasing the number of cardiac ejections per unit time results in increased cardiac output. In addition, increasing the heart rate increases contractility which leads to higher calcium concentrations. With increased heart rate, the time for reuptake of calcium decreases. The increased calcium concentrations cause upregulation of cAMP, which enhances contractility. However, in the setting of myocardial failure, it is not uncommon to observe heart rates high enough that the diastolic interval is shortened and ventricular filling is compromised, resulting in decreased cardiac output. Therefore, rate control becomes paramount in ensuring matched oxygen delivery and utilization in patients with risk for myocardial ischemia.
Myocardial dysfunction can be defined on the basis of perturbed preload, afterload, contractility, or heart rate. Systemic hypovolemia (eg, inadequate preload), secondary to hemorrhage or third-space losses, is the most common etiology in the postsurgical or trauma patient. Other causes of decreased cardiac output may arise from failing or decreased contractile function of the heart. Evolving ischemia can lead to areas of heart muscle that lose their ability to contract, leading to decreased cardiac output. The contractility of heart muscle can also become altered following any insult that results in intrinsic metabolic derangements at the cellular level. This typically occurs in the settings of sepsis, postcardiac arrest, or postcardiopulmonary bypass. Direct physical injury to the myocardium, as occurs following blunt chest injury, may produce contused cardiac muscle, which can lead to contractile dysfunction and decreased cardiac output.
One of the more difficult scenarios to diagnose and manage is right ventricular dysfunction following inferior wall myocardial infarction. Although cardiogenic shock results from right ventricular infarction infrequently, mortality from right ventricular infarction is high (23–53%).7,8 In addition, patients with inferior MI have much higher rates of arrhythmias including atrial fibrillation, ventricular fibrillation and tachycardia and second/third degree AV block.9 The mainstays of treatment include maintaining adequate intravascular volume while supporting a remodeling right ventricle with inotropes.
Pulmonary hypertension may develop after blunt cardiac injury. Most often, this is due to either an acute valvular or papillary disruption, formation of a ventricular septal defect,10 or myocardial damage to the right ventricle. Pulmonary hypertension and right heart failure are difficult to diagnose as there is no particular lab value or routine monitoring that will signal this pathology. Therefore, physical examination findings (new and worsening JVD, a new murmur, unexplained hypotension or hepatic failure, a new arrhythmia, etc) in the proper traumatic setting should prompt further testing with echocardiography. In fact, as many as 27% of major chest trauma patients may have pulmonary hypertension identified on transthoracic echocardiography.11 If this pulmonary hypertension persists beyond the acute traumatic period, it is possible to lead to right heart strain that is also difficult to assess for or make the diagnosis without echocardiography.
Acute traumatic injury to a cardiac valve is rare and is discussed elsewhere (see Chapter 26). Knowledge of common valvular pathology and the impact on trauma resuscitation is important as valve abnormalities can significantly impact the response to fluids, vasopressors and inotropes. Three of the more common valve disorders (aortic stenosis, mitral regurgitation, and tricuspid regurgitation) will be discussed in this section. In aortic stenosis, narrowing of the aortic valve most often leads to symptoms of heart failure with patients complaining of dyspnea on exertion, angina, lightheadedness, or syncope. Persistent and excessive fluid resuscitation and failure of diuresis during recovery may exacerbate the symptoms associated with aortic stenosis. Patients with known aortic stenosis undergoing induction of anesthesia must be ensured adequate preload as rapid hypotension is poorly tolerated due to inadequate compliance of the left ventricle. In addition, similar acute periods of hypotension should be avoided if epidural catheter placement is necessary. Chronic mitral regurgitation may eventually lead to decompensated left ventricular failure. Often, due to left atrial enlargement, these patients suffer from atrial fibrillation. In patients with worsened cardiac function following trauma, offloading the left ventricle by maintaining normotension in patients with mitral regurgitation is key. In addition, responsible fluid management in these patients is necessary as they may also have pulmonary hypertension with excessive fluid resuscitation, leading to the development of pulmonary edema and right heart failure. Acute mitral regurgitation can result from either acute MI or trauma with the rapid development of cardiogenic shock, pulmonary edema, and hemodynamic instability. Given the failure of the mitral valve, the pressures within the left atrium and ventricle will equalize. Treatment includes afterload reduction and emergent surgical repair.12 Tricuspid regurgitation is common but infrequently symptomatic. As with mitral regurgitation, in patients symptomatic (elevated JVD, kidney, and liver dysfunction) from tricuspid regurgitation, fluid overload will lead to right heart failure, then left heart failure and finally, impaired cardiac output with deficits in end organ perfusion.
Due to elevated catecholamines, fluctuations in intravascular volume, electrolyte disturbances, and inflammatory states, critically ill patients are at high risk for the development of arrhythmias.13 The development of an arrhythmia in the ICU is associated with increased mortality with a recent study showing a 17% in-hospital mortality for patients without the development of an arrhythmia, 29% for those who developed a supraventricular arrhythmia, and 73% for those with a ventricular arrhythmia.14 New onset of an arrhythmia in the ICU also leads to increased length of stay by up to 33%.15 The development of any arrhythmia should prompt an evaluation of electrolyte levels, assessment of the rhythm with an ECG and patient assessment for hypotension or signs of impaired perfusion (mental status changes). The management of arrhythmias follows the most recent ACLS guidelines.16
The most common tachyarrhythmia in the critically ill patient is atrial fibrillation.17 If the patient is hemodynamically unstable as evidenced by hypotension and mental status changes, electrical cardioversion is indicated. Medical therapy with β-blockade, calcium channel blockers, or amiodarone can be used to prevent recurrence once the patient has been stabilized. Amiodarone is the preferred agent in patients with hypotension or heart failure.18
Bradyarrhythmias in the critically ill patient can be life-threatening and can be treated with atropine or transcuteneous pacing if there is evidence of hypoperfusion. Most commonly, bradyarrhythmias are caused by elevated intracranial pressure, exaggerated vagal activity, hypothermia, electrolyte abnormalities, and medications (β-blockers, calcium-channel blockers, clonidine, opioids, and dexmedetomidine).8
During the acute resuscitative phase of care, cardiovascular failure in trauma patients is the result of hemorrhage and reversal of this failure occurs only with control of hemorrhage and transfusion of blood products. However, some patients remain in cardiovascular failure despite hemorrhage control and adequate blood product resuscitation. These patients often remain acidotic with large base deficits. The underlying derangement in this circumstance may be cytopathic hypoxia.19,20 Although an infrequent cause of CV failure, it should be considered in the adequately resuscitated but hypotensive and acidotic patient. In fact, cytopathic hypoxia may manifest as profound vasoplegia (see further) with potential etiologies being mitochondrial dysfunction, problems with nitric oxide metabolism, and alterations in hypoxia-induced pathways. Although no specific therapy is available, treatment must be systemic and target all cells (methylene blue, ubiquitin, therapeutic hypothermia, etc).
Myocardial dysfunction secondary to hypovolemia may be caused by hemorrhage or other causes of intravascular volume loss. Prior to the development of hypotension, most adult patients will demonstrate a decrease in urine output, indicative of end-organ hypoperfusion. Augmentation of preload, or restoration of intravascular volume, will reverse this dysfunction if instituted promptly. Normalization of heart rate and blood pressure, along with adequate urine output are simple and effective measurements of adequate volume restoration.
The use of vasopressors during acute, severe traumatic injury resuscitation should be avoided if possible as hemorrhage is the source of almost all shock in the immediate post-injury period (Table 56-1).
| Agent | Typical dosage | HR | MAP | CO | SVR | α+ | B1 | B2 |
|---|---|---|---|---|---|---|---|---|
| Vasopressors: | ||||||||
| Norepinephrine | 0.5–30 μg/min | + | ++ | + | ++ | ++ | + | |
| Vasopressin | 0.04 U/min | ++ | – | ++ | ||||
| Phenylephrine | 0.5–1 μg/kg/min | ++ | ++ | ++ | ||||
| Inotropes: | ||||||||
| Dobutamine | 2–20 μg/kg/min | + | ± | ++ | – | ++ | + | |
| Epinephrine | 0.5–10 μg/min | ++ | ++ | + | ++ | ++ | ++ | ++ |
| Milrinone | 0.125–0.75 μg/kg/min | +++ | – | |||||
| Dopamine | 3–5 μg/kg/min | + | + | ++ | ++ | |||
| 10–20 μg/kg/min | ++ | ++ | + | +++ | ++ | + |
Norepinephrine is an endogenous sympathetic neurotransmitter with α- and β-adrenergic effects. At high doses, α-adrenergic effects predominate, and increased SVR and increased blood pressure result. Specifically, in the setting of right ventricular failure, low-dose norepinephrine improves cardiac function without adversely affecting visceral perfusion.21 Norepinephrine is typically the vasopressor of first choice during or after fluid resuscitation22 and in the setting of sepsis.23 In cardiogenic shock, norepinephrine, when compared to dopamine, is associated with decreased mortality and rates of arrhythmia.24 Norepinephrine is widely used in the care of the head-injured patient in shock because its vasoconstrictive effects do not extend to the cerebral vasculature, making it an ideal agent for maintaining cerebral perfusion pressure.
Vasopressin is a natural hormone produced by the posterior pituitary that acts as a potent vasoconstrictor. Vasopressin is recommended as the second vasopressor during septic shock that is refractory to norepinephrine according to the Surviving Sepsis Campaign.23 When vasopressin is combined with norepinephrine, outcomes in the treatment of catecholamine resistant cardiovascular failure in septic shock are superior to therapy with norepinephrine alone.
Vasopressin has also emerged as a therapy for cardiac arrest and acute resuscitation. Improved neurologic outcomes following cardiac arrest have been shown with the use of vasopressin and steroids in addition to epinephrine in comparison to epinephrine alone.25 In the postoperative cardiotomy patient, vasopressin significantly reduces the need for both pressor and inotropic support, and the development of arrhythmias.26
Phenylephrine is a pure α-agonist and is without any β activity. Phenylephrine is often administered in the bolus form for acute hypotension but may also be infused in patients with hypotension and serious arrhythmias that may be triggered by other vasopressors with β activity.19 Phenylephrine has a duration of action of up to 20 minutes.27 In the setting of cardiogenic shock, phenylephrine is rarely used. At high doses, phenylephrine administration may lead to bradycardia28 and visceral hypoperfusion.
Profound vasoplegia occurs when hypotension and inadequate end organ perfusion persist despite adequate fluid resuscitation, high cardiac output, and the use of multiple vasopressors.29 The exact physiologic mechanism of profound vasoplegia is poorly understood. This profound vasoplegia is most commonly encountered in cardiac surgery patients, especially those who have required cardiopulmonary bypass. Mortality rates, wound infections, days on mechanical ventilation, and lengths of stay are all increased in cardiac patients who have profound vasoplegia.30 Profound vasoplegia may occur in the trauma and postoperative surgical patient as well. Unfortunately, additional treatment modalities for profound vasoplegia are limited. Methylene blue has been used with some success to help to reverse profound vasoplegia. Methylene blue inhibits nitric oxide-mediated vasodilation and can be administered as a slow infusion (2 mg/kg over 20 minutes) for this purpose. Outside of cardiac surgery, methylene blue has been used to reverse vasoplegia in burn patients,31 following liver transplant32 and for septic shock.33 While it should not be considered standard of care, methylene blue may be used in a trauma patient with profound shock unresponsive to fluids or vasopressors as a last resort.
Dobutamine is a synthetic catecholamine with primarily β-adrenegic effects, although it does possess some α1-adrenergic properties. It is primarily an inotrope, increasing contractility, with minimal chronotropic effects. Dobutamine also possesses mild β2-adrenoreceptor activity, producing peripheral vasodilatation. This combination of increased contractility and reduced afterload results in improved cardiac output. Importantly, the increase in cardiac output occurs without an increase in myocardial oxygen consumption.34 Dobutamine may also increase capillary perfusion in septic shock independent of overall hemodynamic effects.35 Because of the vasodilatory effects, dobutamine may reduce blood pressure and is ideally suited for use in low-output cardiac states. For these reasons, dobutamine should be considered the first-choice inotrope for patients with low cardiac output in the presence of adequate preload.36 In addition, dobutamine may be added in the setting of septic shock if cardiac dysfunction is considered to be a contributor to the shock state.
Epinephrine is an endogenous catecholamine with α- and β-adrenergic activity. At low doses, epinephrine exerts primarily β-adrenergic effects, increasing contractility and reducing SVR. Despite this, there is little evidence that epinephrine is superior to dobutamine in the treatment of low-output states. The increases in stroke volume and cardiac output seen with epinephrine have the potential of decreasing blood pressure in patients with inadequate preload. In patients with septic shock that have been adequately fluid resuscitated, epinephrine increases heart rate and stroke volume (and therefore cardiac output) and systemic oxygen delivery without altering vascular tone. Care must be taken when using epinephrine, as renal vasoconstriction, cardiac arrhythmias and splanchnic blood flow,37 and increased myocardial oxygen consumption and demand may result.38 Additionally, metabolic abnormalities are common, including dyskalemias, hyperglycemia, and ketoacidosis.39
Milrinone selectively inhibits phosphodiesterase-3 that leads to increased cAMP and increased calcium intracellularly to promote more efficient contractility. Overall, milrinone administration leads to pulmonary and systemic vasodilation with increased inotropy. Milrinone is used more frequently in the patient with well-established cardiac failure as the half-life is long (2.5 hours) and is not a rapidly titratable medication, unlike dobutamine or epinephrine. As with dobutamine, hypotension may result from either bolus or excessive milrinone use especially in patients who are hypovolemic. In addition, risk of arrhythmia is increased with use of milrinone or dobutamine.
Dopamine is an endogenous catecholamine that has several cardiovascular effects, including increased heart rate, increased contractility, and peripheral vasoconstriction. It is used primarily for inotropic support in order to maintain brain, heart, and kidney perfusion. Dopamine acts on α- and β-adrenoreceptors as well as DA1 and DA2 dopamine receptors, and its actions can be classified based on dose. At doses of 1–3 μg/kg/min, dopamine acts at primarily DA1 receptors in renal, mesenteric, coronary, and cerebral vascular beds, resulting in vasodilation. At moderate doses (3–5 μg/kg/min), dopamine stimulates primarily cardiac β-adrenoreceptor, increasing contractility and thus cardiac output. At higher doses of dopamine (10 μg/kg/min), peripheral vasoconstrictive effects from stimulation of α-adrenergic receptors predominate. This can result in significant coronary vasoconstriction resulting in angina, vasospasm, and increased PAWP. Additionally, increasing afterload from vasoconstriction coupled with an increased heart rate seen at this dose, results in increased myocardial oxygen consumption and demand. Individual variation in the pharmacokinetics of dopamine due to weight-based dosing typically results in poor correlation between blood levels and administered dose. Tachycardia can occur with any dose of dopamine, particularly in the hypovolemic patient. Due to the variable effects of dopamine, the dosage ranges used to define which receptors it affects are to be used as broad guidelines only, with the awareness that low-dose dopamine may have the unwanted effects of medium- or high-dose dopamine on an individual patient. Low-dose dopamine is no longer used for renal protection.40
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


