Fig. 1
IL-6/STAT3 signaling pathway. Shown is a schematic figure of the IL-6/JAK/STAT signaling pathway. Binding of IL-6 to its cognate receptor results in phosphorylation of JAK1/2 with subsequent autophosphorylation of the receptor subunit gp130. Activation of the IL-6 receptor results in recruitment and phosphorylation of the transcription factor STAT3; following its phosphorylation, this transcription factor dimerizes, translocates to the nucleus where it accumulates and regulates gene expression of genes controlling cell survival and proliferation
4.2 Transforming growth factor-β signaling
Transforming growth factor-β (TGF-β) is an important modulator of a broad spectrum of cellular processes, including cellular growth, differentiation, wound repair, and apoptosis. TGF-β ligand binding to its receptor results in the activation of type II (TβRII) and then type I (TβRI) receptors that in turn activate a group of eight ubiquitously expressed globular proteins called Smad-proteins (characterized by homologous regions at their N and C–termini known as Mad homology, MH–1 and MH-2 domains) (Fig. 2) [45−47]. Smad activation results in transcriptional regulation and G1/S suppression of the cell cycle through suppression of Myc, cyclin dependent kinases and others [48, 49]. Further, TGF-β/Smad signaling activates differentiation programs and induces senescence in HCC via upregulation of p21(Cip1) and p15(Ink4b) [50]. TGF-β has a well-established tumor suppressive effect during early carcinogenesis [51]. Inactivation of TGF-β signaling components has been described in hepatobiliary malignancies [52]. The loss of intermediary regulators of TGF-β signaling, such as the Smad3/4 or the adaptor protein β2-Spectrin, result in malignant transformation of stem cells to STICs via loss of differentiation and growth arrest signals and spontaneous HCC formation [5]. Within these liver STICs populations, increased levels of embryonic stem-cell transcription factors Oct4 and Nanog, driven by loss of TGF-β differentiation signals, propagate self-renewal characteristics. In addition to the Smad-protein signaling cascade, TGF-β receptor activation can also activate non-Smad pathways including mitogen-activated protein kinase pathways, such as ERK and p38, as well as the phosphatidylinositol-3-kinase (PI3K)/Akt pathway. These pathways regulate apoptosis, epithelial to mesenchymal transition and cell proliferation. Interestingly, TGF-β is overexpressed in HCC tumor tissue and TGF-β plasma concentrations are elevated in HCC patients [53, 54]. Hence, elevated TGF-β ligand concentrations in the presence of aberrant TGF-βR/Smad signaling could theoretically result in increased signaling of the above described non-Smad pathways, thereby, promoting carcinogenesis and tumor progression.
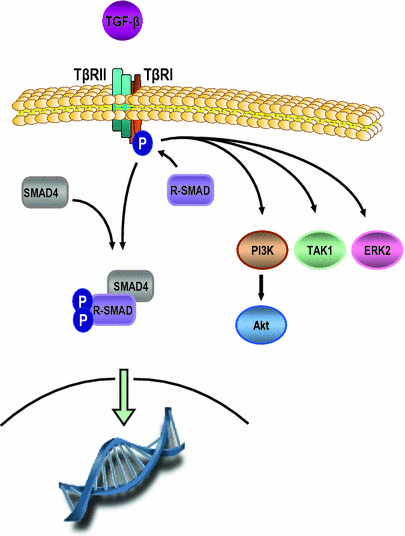
Fig. 2
TGF-β signaling pathway. Shown is the TGF-β signaling pathway. Binding of the TGF-β receptor 2 (TβII) by its TGF-β results in recruitment of TGF-β receptor 1 (TβI) followed by heterodimerization between TβII and TβI, and activation of TβI. Subsequently, receptor-Smad proteins (R-SMAD [i.e. SMAD2/3]) are phosphorylated, oligomerize with co-mediator Smad proteins (i.e. SMAD4) and the complex translocates to the nucleus where it regulates genes controlling tumor suppression. In addition, TGF-β can also activate non-Smad pathways such as PI3K/Akt, ERK and TGF-β activated kinase-1 (TAK1) which can promote cell proliferation
4.3 Wnt/β-Catenin Pathway
The Wnt/β-catenin pathway is an evolutionary conserved pathway regulating tissue cell homeostasis and plays a key role in embryogenesis, tissue regeneration but also carcinogenesis. Wnt ligands compromise a family of soluble cysteine-rich glycoproteins binding to the Frizzled family of cell-surface receptors. In the inactivated state, β-catenin is bound by adenomatosis polyposis coli (APC) and Axin proteins in conjunction with GSK3 resulting in β-catenin phosphorylation and its degradation. Upon ligand binding of the receptors, the downstream effector Dishevelled (DSH) is activated, resulting in inhibition of β-catenin phosphorylation through glycogen synthase kinase-3β (GSK3), thereby, preventing its ubiquitination and proteosomal degradation (Fig. 3). The increasing cytoplasmic concentration of β-catenin is followed by its nuclear translocation and accumulation, where it acts as transcription factor for genes regulating cell proliferation, angiogenesis, extracellular matrix, and apoptosis [55]. Constitutively increased cytoplasmic β-catenin was observed in 62 % and nuclear accumulation in 15 % of HCC tissue samples [56]. β–catenin mutations in HCC with resulting upregulation/accumulation of β-catenin such as the β–Catenin gene CTNNB1 were found in 20–40 % of human HCC, and mutations in its degradation complex such as AXIN1 were found in 3–16 % [57]. Interestingly, in a recent transcriptome meta-analysis, WNT pathway activation in a subset of HCC patients was not simply the result of β-catenin mutation but rather the result of TGF-β activation [58].
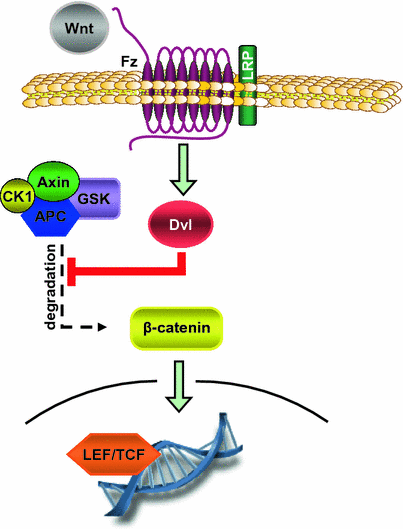
Fig. 3
Canonical Wnt/β-catenin signaling pathway. In the canonical Wnt/β-catenin signaling axis, binding of Wnt ligands to Frizzled receptor (Fz) results in activation in Disheveled (Dvl). Gvl inactivates the Axin/GSK3/CK1/APC complex which physiologically mediates proteosomal degradation of β-catenin. Hence, Wnt/Fz mediates nuclear translocation of β-catenin and gene regulation via inhibition of its degradation. This pathway controls genes regulating cell proliferation, angiogenesis, extracellular matrix, and apoptosis
Dysregulation of the Wnt/β-catenin pathway appears to be an early event in the carcinogenesis of HCC. In particular, it is found in HCV and alcohol associated HCC. Also, β-catenin overexpression has been associated with an increased risk for the development of HCC in hepatic adenomatosis. Interestingly, there is significant evidence of aberrant Wnt/β-catenin pathway signaling in hepatic progenitor cells associated with HCC. In particular, EPCAM has been identified as a target gene of the Wnt-signaling components β-catenin and Tcf4. In human hepatocytes and established HCC cell lines, nuclear accumulation of β-catenin was shown to induce EPCAM while inhibition Tcf4/β-catenin resulted in its downregulation [9]. However, aberrant β-catenin signaling alone is not sufficient for hepatocarcinogenesis but rather requires the concomitant presence of another defect such as the loss of a tumor suppressor gene or activation of a proto-oncogene [59].
4.4 Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) is a key signaling pathway in angiogenesis. It signals through its cognate receptor VEGFR-1, -2, and -3 with the main receptor mediating the VEGF effects being VEGFR-2 (Fig. 4). VEGF is a critical mediator in HCC angiogenesis and largely responsible for abnormal structure and function of liver tumor vessels [60]. VEGF as well as VEGFR1-2 serum concentrations are elevated in HCC patients, and increased expression of VEGF and its receptors is overexpressed in cirrhosis, dysplastic hepatic nodules and HCC [61–63]. Clinically, increased VEGF is associated with poor prognosis in HCC patients [64–66]. Currently, several specific VEGF- and VEGFR-targeted agents are evaluated in phase II trials [60].
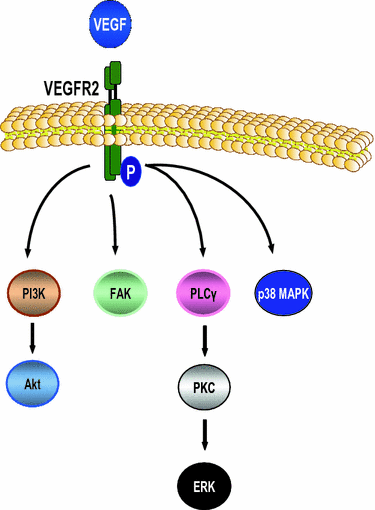
Fig. 4
VEGFR signaling pathway. VEGF signaling is mostly mediated via the EGF receptor 2 (EGFR2). Following binding of ligands of the VEGF family, the receptor is autophosphorylated, internalized and subsequently activates downstream kinase pathways such as PI3K/Akt, focal adhesion kinase (FAK), ERK, and p38 MAPK. This pathway regulates angiogenesis but also other procarcinogenic processes, e.g., cell proliferation
4.5 Insulin-Like Growth Factor
The Insulin-like growth factor (IGF) signaling pathway is activated through high-affinity binding of its ligands IGF-1 and IGF-2 to the IGF-1 receptor (IGF-1R). Following autophosphorylation of IGF-1R, insulin receptor substrate (IRS) and Shc protein mediate activation of the MAPK/MEK/ERK pathways and the PI3K pathway (Fig. 5). The IGF-signaling cascade regulates cell proliferation, mobility and apoptosis. In HCC, IGF-2 is upregulated [67–69]. HBx protein, HCV replication, and aflatoxin induce IGF-2 expression [70–73]. IGF-2 overexpression appears to be an early event in hepatocarcinogenesis based upon animal data using genetically engineered mice constitutively overexpressing IGF-2, and was also observed in the DEN-rat model [74, 75]. Interestingly, the maternally imprinted fetal form of IGF-2 predominates in human HCC tissue in contrast to the expression of the biallelic adult form expressed in non-tumorous hepatic tissue [76]. The IGF-1 receptor is overexpressed in up to 33 % and blockade of this receptor was shown to be a promising therapeutic strategy [77–79]. The functional importance of the activation of the IGF-signaling axis is its procarcinogenic effect, and induction of tumor cell motility and apoptosis resistance mainly via the PI3K/Akt signaling axis [80, 81]. In addition, crosstalk between IGF and EGF-signaling has been proposed as a mechanism of EGFR-resistance in HCC [77, 82]. Preclinical in vitro and in vivo data indicate a therapeutic benefit through a IGF-inhibition strategy through induction of apoptosis and inhibition of cell proliferation [83].
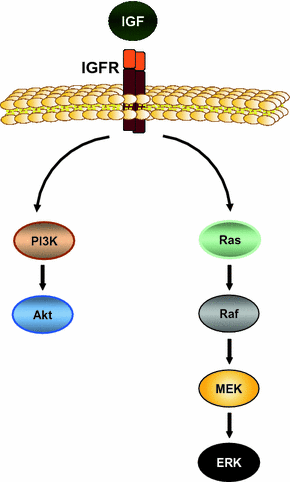
Fig. 5
IGF-signaling pathway. The IGF-signaling pathway is activated by binding of the ligands IGF-1 and IGF-2 to their cognate receptor IGF-1R. Ligand binding results in autophosphorylation of the receptor followed by activation of its downstream effectors such as PI3K/Akt and Ras/Raf/MEK/ERK signaling cascade. Thereby, IGF regulates several cellular processes such as proliferation, cell motility, and apoptosis
4.6 Hepatocyte Growth Factor/c-Met
Hepatocyte Growth factor (HGF) is a growth factor normally expressed and secreted by stromal cells and shown to play a key role in liver regeneration, hepatocyte survival, and tissue remodeling after acute injury [84, 85]. It activates the receptor c-Met followed by phosphorylation of its adaptor protein complex consisting of GRB2 and GAB1 (Fig. 6). Subsequently, the downstream targets PI3K and ERK are activated resulting in regulation of genes driving cell proliferation, cell motility, cell–cell dissociation and survival [86]. HGF/c-Met in carcinogenesis mediates proliferation, invasion, and metastases formation [86, 87]. C-Met is overexpressed in up to 48 % of human HCC and correlated to with an aggressive HCC phenotype and the prognosis of HCC patients [88, 89]. In vitro, c-Met overexpressing HCC cells displayed stem cell characteristics including chemoresistance; treatment of these c-Met positive cells in vitro and in a subcutaneous xenograft murine model with a targeted c-Met tyrosine kinase inhibitor resulted in decreased tumor cell proliferation and tumor growth [90].
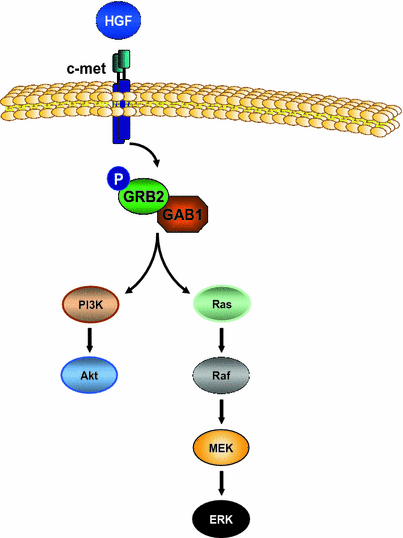
Fig. 6
HGF/c-Met signaling pathway. Shown is a schematic figure of the HGF/c-Met signaling pathway. c-Met binding by HGF results in autophosphorylation of c-Met followed by activation of the adaptor protein complex GRB2/GAB1. Subsequently, the PI3K/Akt and Ras/Raf/MEK/ERK signaling cascades are activated with resulting regulation of genes driving cell proliferation, cell motility, cell–cell dissociation, and survival
4.7 Raf/MEK/ERK
The cytosolic kinases Raf, MEK, and ERK are downstream of the above described receptor kinases. Phosphorylation of receptor kinases is followed by activation of the adaptor molecule complex GRB2/SHC/SOS which in turn activates Raf/MEK/ERK. Within this cascade, the GTP-ase Ras and the serine/threonine protein kinase Raf (a-Raf, b-Raf, c-Raf) are the main signaling regulators activating downstream the mitogen/extracellular protein kinases (MAP2K) MEK1 and MEK1. MEK1/2 activate downstream the mitogen/extracellular protein kinase kinases (MAP3K) ERK1 and ERK2. ERK1/2 regulate multiple cell functions through different cytosolic and nuclear inducers of gene expression (i.e. transcription factors, cell-cycle related kinases) and interaction with other signaling pathways [55]. This signaling axis regulates cellular processes such as proliferation, differentiation, angiogenesis, and survival, and plays a key role in malignancies including HCC [91]. Enhanced signaling through this pathway can either be mediated via upstream kinases, activating mutations of components of this pathway or external factors. HCV core protein was shown to activate c-Raf, MEK1, and ERK1/2 [92]. Raf/MEK/ERK are constitutively activated in HCC and c-Raf overexpression was found in 100 % of human HCC samples [93, 94]. In vitro data also indicate the potential of the HBV protein HBx to induce the overexpression of c-Raf [95]. The -Raf inhibitory proteins Raf-1 kinase inhibitory protein (RKIP) and KRAS and BRAF mutations are extremely rare in HCC [96].
4.8 Phosphatidylinositol 3-kinase/Akt/mTOR
Phosphatidylinositol 3-kinase (PI3K) is the downstream target of several receptor kinase pathways including the IL-6, TGF-β, EGF, and IGF. Upon its activation, the second messenger phosphoinositoltriphosphate (PIP3b) activates the serine/threonine kinase Akt. Activated Akt activates transcription factors (i.e. FOXO) and phosphorylates the cytoplasmatic proteins mTOR and the Bcl-2 associated death promoter (BAD), thereby, increasing cell proliferation and decreasing apoptosis. Physiologically, this pathway is controlled by phosphatases and tensin homolog (PTEN) through dephosphorylation of PI3K regulated proteins [55]. As outlined above, the majority of the PI3K-activating upstream pathways are up-regulated in HCC (i.e. IL-6, IGF). Phosphorylated mTOR levels were found to be increased in 15 % and the levels of the activated Akt substrate p70 S6 kinase in 45 % [97]. The negative regulator PTEN gene is mutated in 5 % and the PTEN protein expression decreased or absent in 41 % of HCC; clinically, loss of PTEN was associated with tumor progression and poor prognosis [98]. Similarly, positive immunohistochemical positivity for phosphorylated Akt was a poor prognostic factor for HCC recurrence on multivariate analysis [99]. The importance of this pathway in carcinogenesis and tumor progression was further confirmed in animal studies using a albumin-regulated PTEN knockout mouse model in which 66 % of animals developed HCC [100]. The potential of Akt- or mTOR-inhibitors in the treatment or prevention of HCC has been shown in preclinical and few clinical trials [101]; however, further randomized controlled trails are needed to assess the therapeutic potential of an Akt/mTOR-targeted strategy.
5 Summary
HCC is one of the leading causes of cancer-related mortality worldwide. Its incidence is increasing, in particular in Western industrial nations. In the last three decades, the mortality of this malignancy has not changed significantly. Our knowledge of the molecular carcinogenesis and the signaling pathways promoting the progression, chemoresistance, and cell migration of this malignancy has considerably grown. This development resulted in the development of novel, molecular-targeted agents such as sorafenib shown to improve survival of HCC patients [102]. However, the survival benefit is marginal with 3 months. Recently, more evidences have been collected about STICs as tumor initiating cells and dysregulated pathways promoting their apoptosis resistance. In particular, IL-6/STAT3 and TGF-β signaling have been shown to be key signaling pathways with a great potential as therapeutic targets.
References
1.
2.
3.
4.
5.
Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, Ressom HW, Rashid A, He AR, Mendelson JS, Jessup JM, Shetty K, Zasloff M, Mishra B, Reddy EP, Johnson L, Mishra L (2008) Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A 105:2445–50PubMedCrossRef
6.
7.
Furuyama K, Kawaguchi Y et al. (2011) Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43:34–41
8.
9.
Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, Reid LM, Minato H, Honda M, Kaneko S, Tang ZY, Wang XW (2009) EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 136:1012–24PubMedCrossRef
10.
Lee JS, Heo J et al. (2006) A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med, 12:410–16
11.
Marquardt JU, Raggi C et al. (2011) Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology, 54: 1031–1042
12.
Ji J, Yamashita T, Budhu A, Forgues M, Jia HL, Li C, Deng C, Wauthier E, Reid LM, Ye QH, Qin LX, Yang W, Wang HY, Tang ZY, Croce CM, Wang XW (2009) Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology 50:472–80PubMedCrossRef
13.
14.
15.
16.
Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G (2008) The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48:1224–31PubMedCrossRef
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


